sceptre is an R package for single-cell CRISPR screen data analysis,
emphasizing statistical rigor, massive scalability, and ease of use.
Author
Maintainer: Timothy Barry tbarry@hsph.harvard.edu (ORCID)
Authors:
Louis Deutsch
Eugene Katsevich ekatsevi@wharton.upenn.edu
Other contributors:
Wharton Analytics [funder]
National Science Foundation (Grants DMS-2113072 and DMS-2310654) [funder]
Examples
##########################
# Low-MOI CRISPRko example
##########################
# 1. create the sceptre object
data("lowmoi_example_data")
sceptre_object <- import_data(
response_matrix = lowmoi_example_data$response_matrix,
grna_matrix = lowmoi_example_data$grna_matrix,
extra_covariates = lowmoi_example_data$extra_covariates,
grna_target_data_frame = lowmoi_example_data$grna_target_data_frame,
moi = "low"
)
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 1000 cells
#> • 100 responses
#> • Low multiplicity-of-infection
#> • 40 targeting gRNAs (distributed across 20 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✗ set_analysis_parameters()
#> ✗ assign_grnas()
#> ✗ run_qc()
#> ✗ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: not specified
#> • Positive control pairs: not specified
#> • Sidedness of test: not specified
#> • Control group: not specified
#> • Resampling mechanism: not specified
#> • gRNA integration strategy: not specified
#> • Resampling approximation: not specified
#> • Multiple testing adjustment: none
#> • N nonzero treatment cells threshold: not specified
#> • N nonzero control cells threshold: not specified
#> • Formula object: not specified
# 2. set the analysis parameters
positive_control_pairs <- construct_positive_control_pairs(sceptre_object)
discovery_pairs <- construct_trans_pairs(
sceptre_object = sceptre_object,
positive_control_pairs = positive_control_pairs,
pairs_to_exclude = "pc_pairs"
)
sceptre_object <- set_analysis_parameters(
sceptre_object = sceptre_object,
discovery_pairs = discovery_pairs,
positive_control_pairs = positive_control_pairs
)
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 1000 cells
#> • 100 responses
#> • Low multiplicity-of-infection
#> • 40 targeting gRNAs (distributed across 20 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✗ assign_grnas()
#> ✗ run_qc()
#> ✗ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1980 pairs
#> • Positive control pairs: data frame with 20 pairs
#> • Sidedness of test: both
#> • Control group: non-targeting cells
#> • Resampling mechanism: permutations
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: not specified
#> • N nonzero control cells threshold: not specified
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + batch
# 3. assign grnas
plot_grna_count_distributions(sceptre_object)
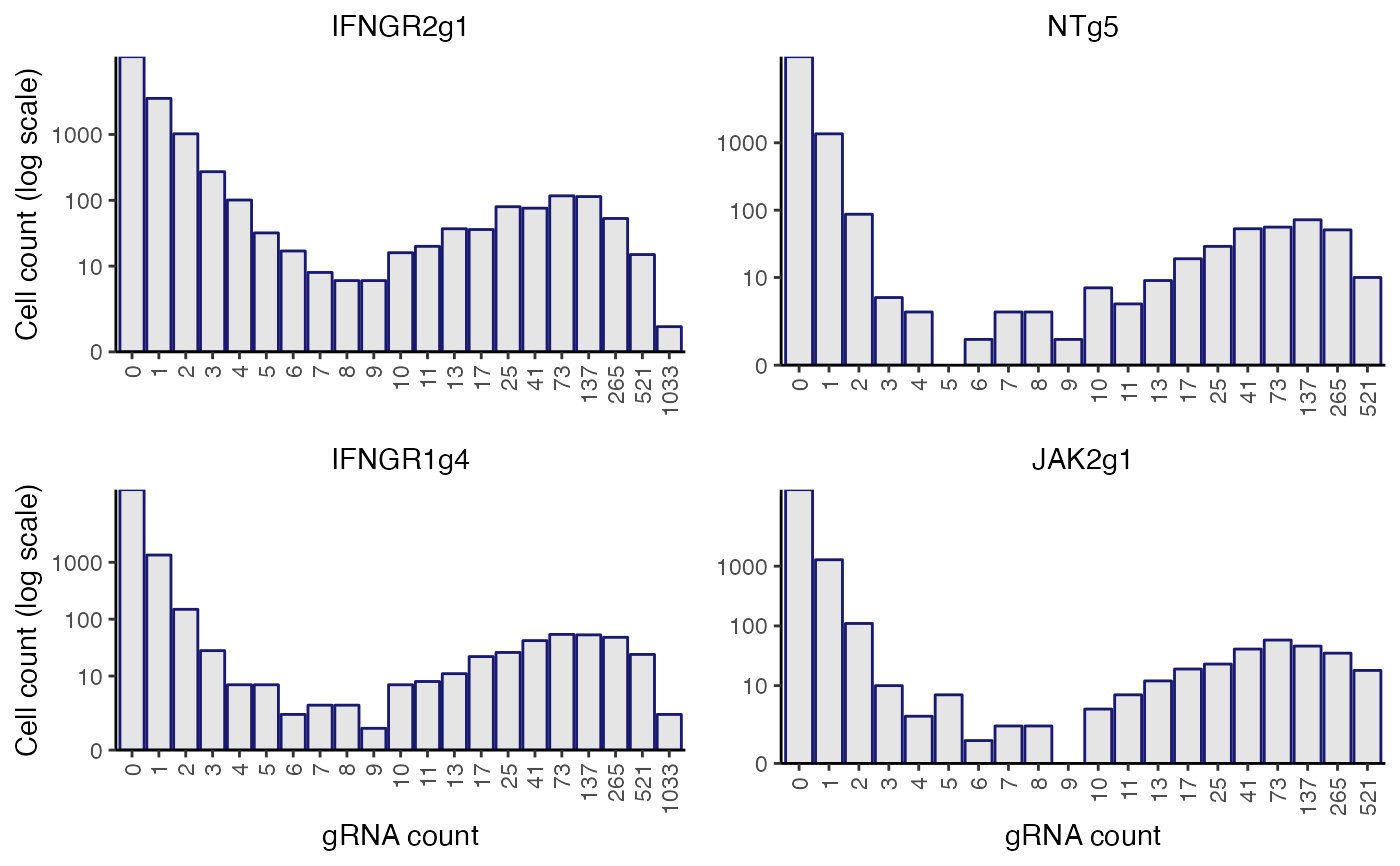 sceptre_object <- sceptre_object |> assign_grnas()
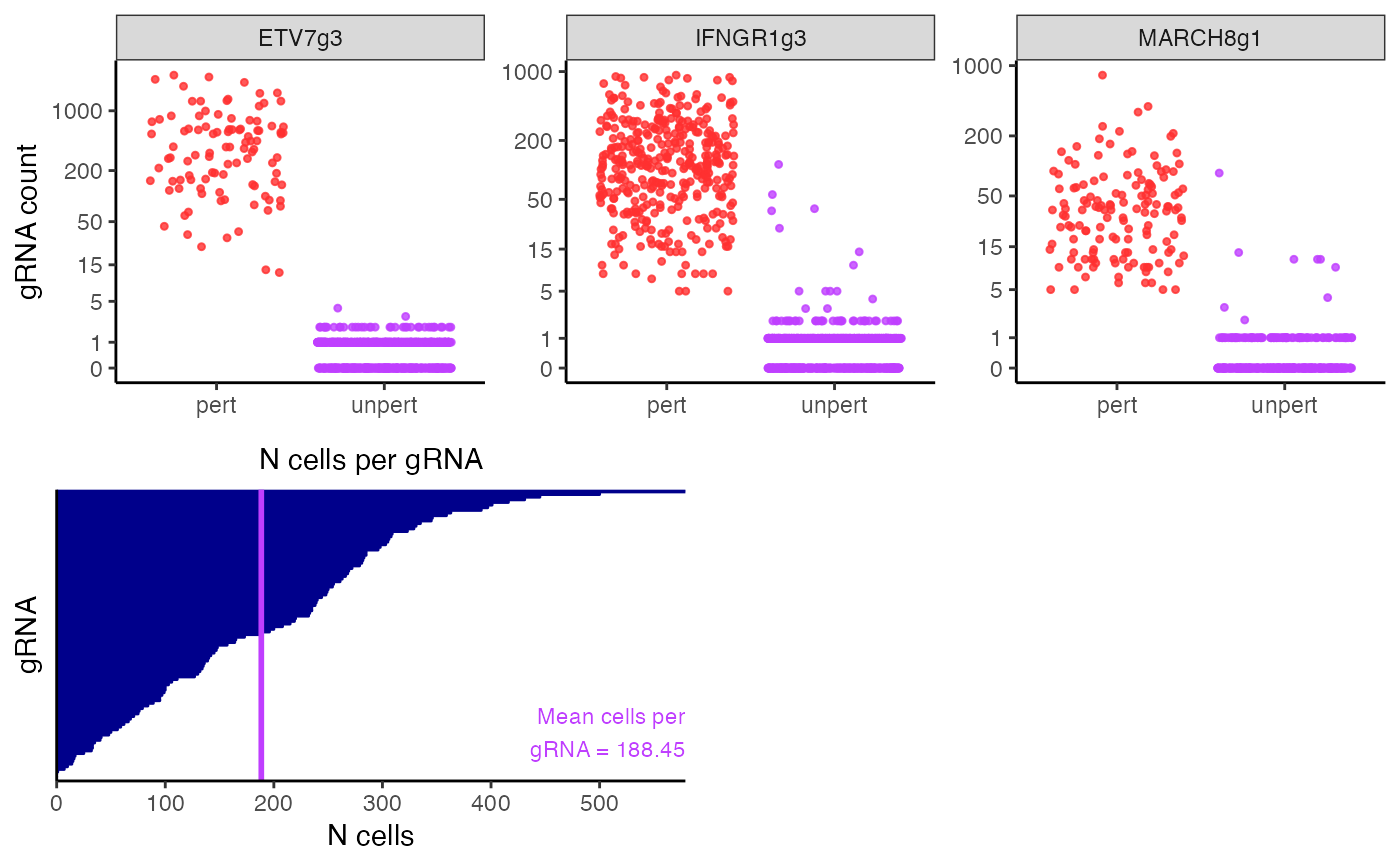
plot(sceptre_object)
sceptre_object <- sceptre_object |> assign_grnas()
plot(sceptre_object)
 print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 1000 cells
#> • 100 responses
#> • Low multiplicity-of-infection
#> • 40 targeting gRNAs (distributed across 20 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✗ run_qc()
#> ✗ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1980 pairs
#> • Positive control pairs: data frame with 20 pairs
#> • Sidedness of test: both
#> • Control group: non-targeting cells
#> • Resampling mechanism: permutations
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: not specified
#> • N nonzero control cells threshold: not specified
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: maximum
#> • Mean N cells per gRNA: 20
#> • Mean N gRNAs per cell (MOI): not computed when using "maximum" assignment method
# 4. run qc
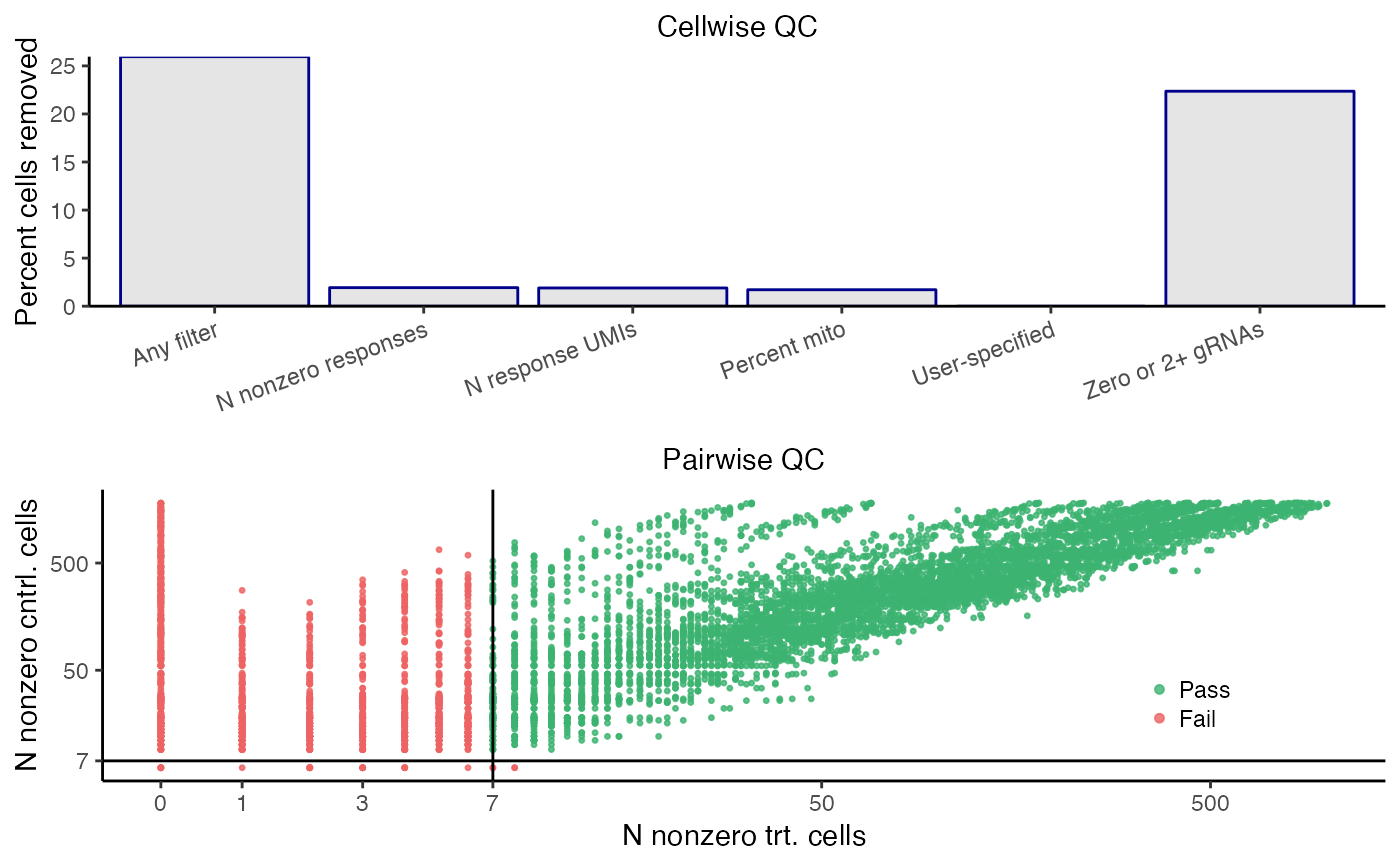
plot_covariates(sceptre_object, p_mito_threshold = 0.075)
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 1000 cells
#> • 100 responses
#> • Low multiplicity-of-infection
#> • 40 targeting gRNAs (distributed across 20 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✗ run_qc()
#> ✗ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1980 pairs
#> • Positive control pairs: data frame with 20 pairs
#> • Sidedness of test: both
#> • Control group: non-targeting cells
#> • Resampling mechanism: permutations
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: not specified
#> • N nonzero control cells threshold: not specified
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: maximum
#> • Mean N cells per gRNA: 20
#> • Mean N gRNAs per cell (MOI): not computed when using "maximum" assignment method
# 4. run qc
plot_covariates(sceptre_object, p_mito_threshold = 0.075)
 sceptre_object <- sceptre_object |> run_qc(p_mito_threshold = 0.075)
plot(sceptre_object)
sceptre_object <- sceptre_object |> run_qc(p_mito_threshold = 0.075)
plot(sceptre_object)
 print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 1000 cells (811 after cellwise QC)
#> • 100 responses
#> • Low multiplicity-of-infection
#> • 40 targeting gRNAs (distributed across 20 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✗ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1980 pairs (1911 after pairwise QC)
#> • Positive control pairs: data frame with 20 pairs (11 after pairwise QC)
#> • Sidedness of test: both
#> • Control group: non-targeting cells
#> • Resampling mechanism: permutations
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: maximum
#> • Mean N cells per gRNA: 20
#> • Mean N gRNAs per cell (MOI): not computed when using "maximum" assignment method
# 5. run the calibration check
sceptre_object <- run_calibration_check(sceptre_object)
#> Note: If you are on a Mac laptop or desktop, consider setting `parallel = TRUE` to improve speed. Otherwise, keep `parallel = FALSE`.
#> Constructing negative control pairs.
#> ✓
#> Generating permutation resamples.
#> ✓
#> Analyzing pairs containing response ENSG00000234860 (1 of 100)
#> Analyzing pairs containing response ENSG00000148482 (5 of 100)
#> Analyzing pairs containing response ENSG00000230789 (10 of 100)
#> Analyzing pairs containing response ENSG00000100122 (15 of 100)
#> Analyzing pairs containing response ENSG00000285699 (20 of 100)
#> Analyzing pairs containing response ENSG00000253141 (25 of 100)
#> Analyzing pairs containing response ENSG00000154803 (30 of 100)
#> Analyzing pairs containing response ENSG00000287671 (35 of 100)
#> Analyzing pairs containing response ENSG00000178199 (40 of 100)
#> Analyzing pairs containing response ENSG00000221937 (45 of 100)
#> Analyzing pairs containing response ENSG00000235335 (50 of 100)
#> Analyzing pairs containing response ENSG00000169992 (55 of 100)
#> Analyzing pairs containing response ENSG00000228008 (60 of 100)
#> Analyzing pairs containing response ENSG00000233251 (65 of 100)
#> Analyzing pairs containing response ENSG00000166482 (70 of 100)
#> Analyzing pairs containing response ENSG00000259269 (75 of 100)
#> Analyzing pairs containing response ENSG00000267009 (80 of 100)
#> Analyzing pairs containing response ENSG00000143314 (85 of 100)
#> Analyzing pairs containing response ENSG00000234580 (90 of 100)
#> Analyzing pairs containing response ENSG00000136997 (95 of 100)
#> Analyzing pairs containing response ENSG00000251187 (100 of 100)
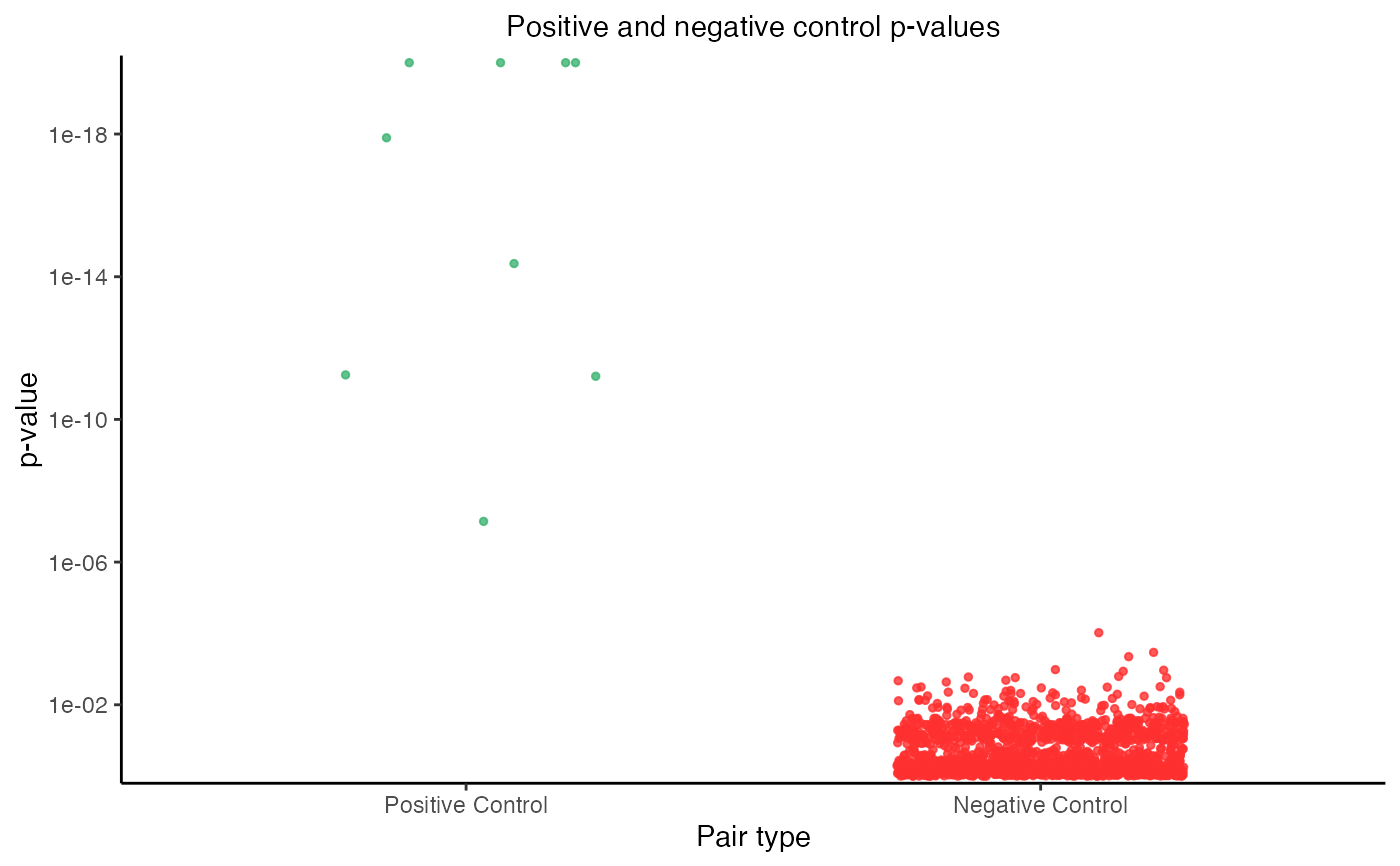
plot(sceptre_object)
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 1000 cells (811 after cellwise QC)
#> • 100 responses
#> • Low multiplicity-of-infection
#> • 40 targeting gRNAs (distributed across 20 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✗ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1980 pairs (1911 after pairwise QC)
#> • Positive control pairs: data frame with 20 pairs (11 after pairwise QC)
#> • Sidedness of test: both
#> • Control group: non-targeting cells
#> • Resampling mechanism: permutations
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: maximum
#> • Mean N cells per gRNA: 20
#> • Mean N gRNAs per cell (MOI): not computed when using "maximum" assignment method
# 5. run the calibration check
sceptre_object <- run_calibration_check(sceptre_object)
#> Note: If you are on a Mac laptop or desktop, consider setting `parallel = TRUE` to improve speed. Otherwise, keep `parallel = FALSE`.
#> Constructing negative control pairs.
#> ✓
#> Generating permutation resamples.
#> ✓
#> Analyzing pairs containing response ENSG00000234860 (1 of 100)
#> Analyzing pairs containing response ENSG00000148482 (5 of 100)
#> Analyzing pairs containing response ENSG00000230789 (10 of 100)
#> Analyzing pairs containing response ENSG00000100122 (15 of 100)
#> Analyzing pairs containing response ENSG00000285699 (20 of 100)
#> Analyzing pairs containing response ENSG00000253141 (25 of 100)
#> Analyzing pairs containing response ENSG00000154803 (30 of 100)
#> Analyzing pairs containing response ENSG00000287671 (35 of 100)
#> Analyzing pairs containing response ENSG00000178199 (40 of 100)
#> Analyzing pairs containing response ENSG00000221937 (45 of 100)
#> Analyzing pairs containing response ENSG00000235335 (50 of 100)
#> Analyzing pairs containing response ENSG00000169992 (55 of 100)
#> Analyzing pairs containing response ENSG00000228008 (60 of 100)
#> Analyzing pairs containing response ENSG00000233251 (65 of 100)
#> Analyzing pairs containing response ENSG00000166482 (70 of 100)
#> Analyzing pairs containing response ENSG00000259269 (75 of 100)
#> Analyzing pairs containing response ENSG00000267009 (80 of 100)
#> Analyzing pairs containing response ENSG00000143314 (85 of 100)
#> Analyzing pairs containing response ENSG00000234580 (90 of 100)
#> Analyzing pairs containing response ENSG00000136997 (95 of 100)
#> Analyzing pairs containing response ENSG00000251187 (100 of 100)
plot(sceptre_object)
 print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 1000 cells (811 after cellwise QC)
#> • 100 responses
#> • Low multiplicity-of-infection
#> • 40 targeting gRNAs (distributed across 20 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✓ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1980 pairs (1911 after pairwise QC)
#> • Positive control pairs: data frame with 20 pairs (11 after pairwise QC)
#> • Sidedness of test: both
#> • Control group: non-targeting cells
#> • Resampling mechanism: permutations
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: maximum
#> • Mean N cells per gRNA: 20
#> • Mean N gRNAs per cell (MOI): not computed when using "maximum" assignment method
#>
#> Summary of results:
#> • N negative control pairs called as significant: 0/1911
#> • Mean log-2 FC for negative control pairs: -0.03
# 6. run power check
sceptre_object <- run_power_check(sceptre_object)
#> Note: If you are on a Mac laptop or desktop, consider setting `parallel = TRUE` to improve speed. Otherwise, keep `parallel = FALSE`.
#> Generating permutation resamples.
#> ✓
#> Analyzing pairs containing response ENSG00000287679 (1 of 11)
#> Analyzing pairs containing response ENSG00000147324 (5 of 11)
#> Analyzing pairs containing response ENSG00000181374 (10 of 11)
plot(sceptre_object)
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 1000 cells (811 after cellwise QC)
#> • 100 responses
#> • Low multiplicity-of-infection
#> • 40 targeting gRNAs (distributed across 20 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✓ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1980 pairs (1911 after pairwise QC)
#> • Positive control pairs: data frame with 20 pairs (11 after pairwise QC)
#> • Sidedness of test: both
#> • Control group: non-targeting cells
#> • Resampling mechanism: permutations
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: maximum
#> • Mean N cells per gRNA: 20
#> • Mean N gRNAs per cell (MOI): not computed when using "maximum" assignment method
#>
#> Summary of results:
#> • N negative control pairs called as significant: 0/1911
#> • Mean log-2 FC for negative control pairs: -0.03
# 6. run power check
sceptre_object <- run_power_check(sceptre_object)
#> Note: If you are on a Mac laptop or desktop, consider setting `parallel = TRUE` to improve speed. Otherwise, keep `parallel = FALSE`.
#> Generating permutation resamples.
#> ✓
#> Analyzing pairs containing response ENSG00000287679 (1 of 11)
#> Analyzing pairs containing response ENSG00000147324 (5 of 11)
#> Analyzing pairs containing response ENSG00000181374 (10 of 11)
plot(sceptre_object)
 print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 1000 cells (811 after cellwise QC)
#> • 100 responses
#> • Low multiplicity-of-infection
#> • 40 targeting gRNAs (distributed across 20 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✓ run_calibration_check()
#> ✓ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1980 pairs (1911 after pairwise QC)
#> • Positive control pairs: data frame with 20 pairs (11 after pairwise QC)
#> • Sidedness of test: both
#> • Control group: non-targeting cells
#> • Resampling mechanism: permutations
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: maximum
#> • Mean N cells per gRNA: 20
#> • Mean N gRNAs per cell (MOI): not computed when using "maximum" assignment method
#>
#> Summary of results:
#> • N negative control pairs called as significant: 0/1911
#> • Mean log-2 FC for negative control pairs: -0.03
#> • Median positive control p-value: 0.00048
# 7. run discovery analysis
sceptre_object <- run_discovery_analysis(sceptre_object)
#> Note: If you are on a Mac laptop or desktop, consider setting `parallel = TRUE` to improve speed. Otherwise, keep `parallel = FALSE`.
#> Generating permutation resamples.
#> ✓
#> Analyzing pairs containing response ENSG00000230714 (1 of 100)
#> Analyzing pairs containing response ENSG00000258818 (5 of 100)
#> Analyzing pairs containing response ENSG00000178038 (10 of 100)
#> Analyzing pairs containing response ENSG00000261469 (15 of 100)
#> Analyzing pairs containing response ENSG00000164466 (20 of 100)
#> Analyzing pairs containing response ENSG00000259221 (25 of 100)
#> Analyzing pairs containing response ENSG00000181803 (30 of 100)
#> Analyzing pairs containing response ENSG00000266651 (35 of 100)
#> Analyzing pairs containing response ENSG00000267788 (40 of 100)
#> Analyzing pairs containing response ENSG00000169992 (45 of 100)
#> Analyzing pairs containing response ENSG00000166482 (50 of 100)
#> Analyzing pairs containing response ENSG00000105122 (55 of 100)
#> Analyzing pairs containing response ENSG00000271855 (60 of 100)
#> Analyzing pairs containing response ENSG00000251187 (65 of 100)
#> Analyzing pairs containing response ENSG00000169836 (70 of 100)
#> Analyzing pairs containing response ENSG00000131016 (75 of 100)
#> Analyzing pairs containing response ENSG00000198851 (80 of 100)
#> Analyzing pairs containing response ENSG00000211767 (85 of 100)
#> Analyzing pairs containing response ENSG00000143314 (90 of 100)
#> Analyzing pairs containing response ENSG00000181374 (95 of 100)
#> Analyzing pairs containing response ENSG00000260003 (100 of 100)
plot(sceptre_object)
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 1000 cells (811 after cellwise QC)
#> • 100 responses
#> • Low multiplicity-of-infection
#> • 40 targeting gRNAs (distributed across 20 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✓ run_calibration_check()
#> ✓ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1980 pairs (1911 after pairwise QC)
#> • Positive control pairs: data frame with 20 pairs (11 after pairwise QC)
#> • Sidedness of test: both
#> • Control group: non-targeting cells
#> • Resampling mechanism: permutations
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: maximum
#> • Mean N cells per gRNA: 20
#> • Mean N gRNAs per cell (MOI): not computed when using "maximum" assignment method
#>
#> Summary of results:
#> • N negative control pairs called as significant: 0/1911
#> • Mean log-2 FC for negative control pairs: -0.03
#> • Median positive control p-value: 0.00048
# 7. run discovery analysis
sceptre_object <- run_discovery_analysis(sceptre_object)
#> Note: If you are on a Mac laptop or desktop, consider setting `parallel = TRUE` to improve speed. Otherwise, keep `parallel = FALSE`.
#> Generating permutation resamples.
#> ✓
#> Analyzing pairs containing response ENSG00000230714 (1 of 100)
#> Analyzing pairs containing response ENSG00000258818 (5 of 100)
#> Analyzing pairs containing response ENSG00000178038 (10 of 100)
#> Analyzing pairs containing response ENSG00000261469 (15 of 100)
#> Analyzing pairs containing response ENSG00000164466 (20 of 100)
#> Analyzing pairs containing response ENSG00000259221 (25 of 100)
#> Analyzing pairs containing response ENSG00000181803 (30 of 100)
#> Analyzing pairs containing response ENSG00000266651 (35 of 100)
#> Analyzing pairs containing response ENSG00000267788 (40 of 100)
#> Analyzing pairs containing response ENSG00000169992 (45 of 100)
#> Analyzing pairs containing response ENSG00000166482 (50 of 100)
#> Analyzing pairs containing response ENSG00000105122 (55 of 100)
#> Analyzing pairs containing response ENSG00000271855 (60 of 100)
#> Analyzing pairs containing response ENSG00000251187 (65 of 100)
#> Analyzing pairs containing response ENSG00000169836 (70 of 100)
#> Analyzing pairs containing response ENSG00000131016 (75 of 100)
#> Analyzing pairs containing response ENSG00000198851 (80 of 100)
#> Analyzing pairs containing response ENSG00000211767 (85 of 100)
#> Analyzing pairs containing response ENSG00000143314 (90 of 100)
#> Analyzing pairs containing response ENSG00000181374 (95 of 100)
#> Analyzing pairs containing response ENSG00000260003 (100 of 100)
plot(sceptre_object)
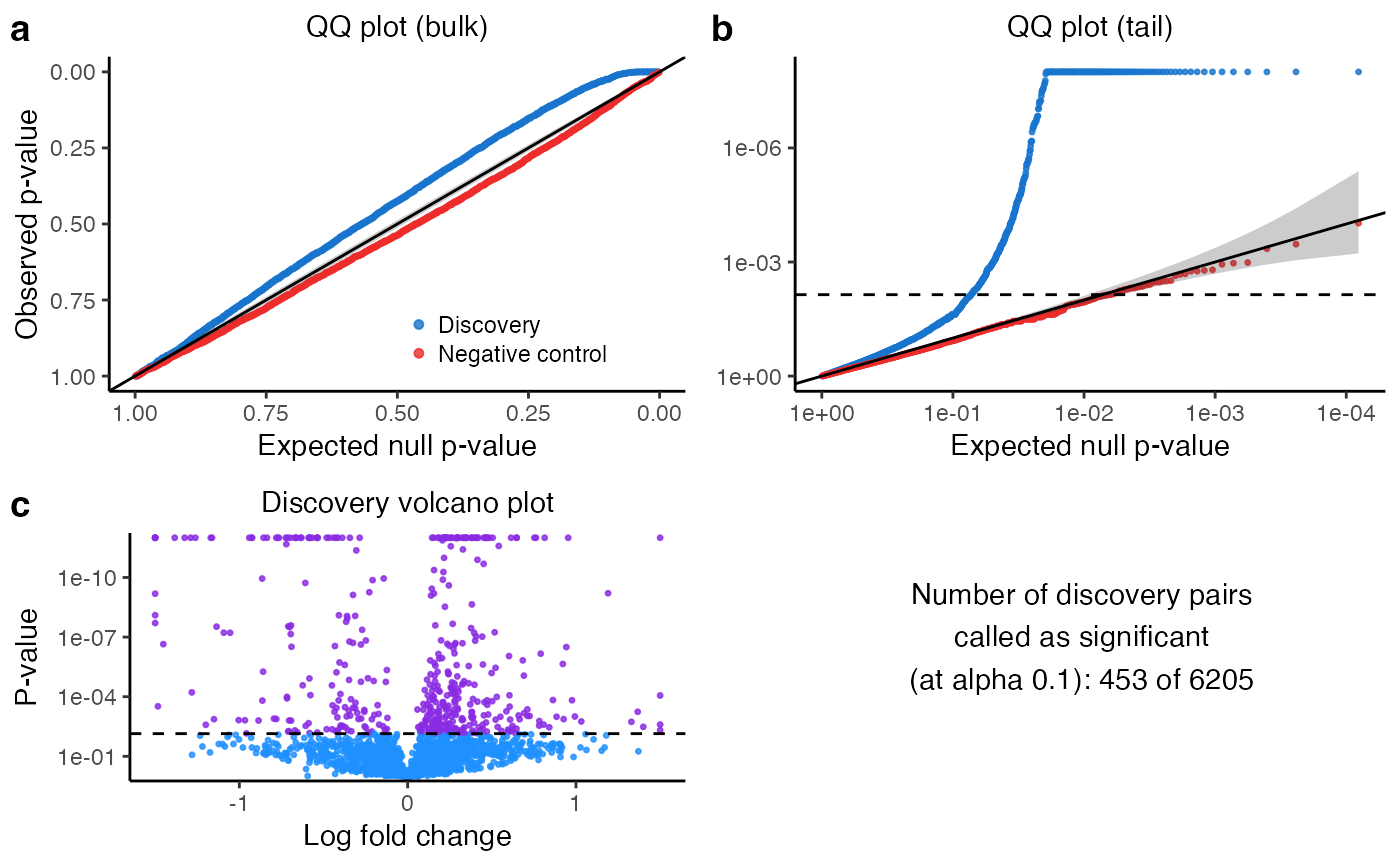 print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 1000 cells (811 after cellwise QC)
#> • 100 responses
#> • Low multiplicity-of-infection
#> • 40 targeting gRNAs (distributed across 20 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✓ run_calibration_check()
#> ✓ run_power_check()
#> ✓ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1980 pairs (1911 after pairwise QC)
#> • Positive control pairs: data frame with 20 pairs (11 after pairwise QC)
#> • Sidedness of test: both
#> • Control group: non-targeting cells
#> • Resampling mechanism: permutations
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: maximum
#> • Mean N cells per gRNA: 20
#> • Mean N gRNAs per cell (MOI): not computed when using "maximum" assignment method
#>
#> Summary of results:
#> • N negative control pairs called as significant: 0/1911
#> • Mean log-2 FC for negative control pairs: -0.03
#> • Median positive control p-value: 0.00048
#> • N discovery pairs called as significant: 37/1911
# 8. write results to a directory. tempdir() is used so this example is
# self-contained; for a real analysis choose a directory you can find again.
output_dir <- file.path(tempdir(), "sceptre_outputs_lowmoi")
write_outputs_to_directory(
sceptre_object = sceptre_object,
directory = output_dir
)
message(
"sceptre outputs written to a temporary directory; ",
'open with `browseURL("', output_dir, '")`'
)
#> sceptre outputs written to a temporary directory; open with `browseURL("/var/folders/1w/h831hyps5qs5lzkh5xjj0_wh0000gq/T//RtmpgcetiN/sceptre_outputs_lowmoi")`
##########################
# High-MOI CRISPRi example
##########################
# 1. create the sceptre object from cellranger output
directories <- paste0(
system.file("extdata", package = "sceptre"),
"/highmoi_example/gem_group_", c(1, 2)
)
data(grna_target_data_frame_highmoi)
sceptre_object <- import_data_from_cellranger(
directories = directories,
moi = "high",
grna_target_data_frame = grna_target_data_frame_highmoi
)
#> Processing directory 1.
#> ✓
#> Processing directory 2.
#> ✓
#> Combining matrices across directories.
#> ✓
#> Creating the sceptre object.
#> ✓
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 500 cells
#> • 100 responses
#> • High multiplicity-of-infection
#> • 50 targeting gRNAs (distributed across 25 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✗ set_analysis_parameters()
#> ✗ assign_grnas()
#> ✗ run_qc()
#> ✗ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: not specified
#> • Positive control pairs: not specified
#> • Sidedness of test: not specified
#> • Resampling mechanism: not specified
#> • gRNA integration strategy: not specified
#> • Resampling approximation: not specified
#> • Multiple testing adjustment: none
#> • N nonzero treatment cells threshold: not specified
#> • N nonzero control cells threshold: not specified
#> • Formula object: not specified
# 2. set the analysis parameters
positive_control_pairs <- construct_positive_control_pairs(sceptre_object)
discovery_pairs <- construct_cis_pairs(sceptre_object,
positive_control_pairs = positive_control_pairs,
distance_threshold = 5e6
)
sceptre_object <- set_analysis_parameters(
sceptre_object = sceptre_object,
discovery_pairs = discovery_pairs,
positive_control_pairs = positive_control_pairs,
side = "left"
)
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 500 cells
#> • 100 responses
#> • High multiplicity-of-infection
#> • 50 targeting gRNAs (distributed across 25 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✗ assign_grnas()
#> ✗ run_qc()
#> ✗ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1406 pairs
#> • Positive control pairs: data frame with 5 pairs
#> • Sidedness of test: left
#> • Resampling mechanism: conditional resampling
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: not specified
#> • N nonzero control cells threshold: not specified
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
# 3. assign grnas
plot_grna_count_distributions(sceptre_object)
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 1000 cells (811 after cellwise QC)
#> • 100 responses
#> • Low multiplicity-of-infection
#> • 40 targeting gRNAs (distributed across 20 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✓ run_calibration_check()
#> ✓ run_power_check()
#> ✓ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1980 pairs (1911 after pairwise QC)
#> • Positive control pairs: data frame with 20 pairs (11 after pairwise QC)
#> • Sidedness of test: both
#> • Control group: non-targeting cells
#> • Resampling mechanism: permutations
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: maximum
#> • Mean N cells per gRNA: 20
#> • Mean N gRNAs per cell (MOI): not computed when using "maximum" assignment method
#>
#> Summary of results:
#> • N negative control pairs called as significant: 0/1911
#> • Mean log-2 FC for negative control pairs: -0.03
#> • Median positive control p-value: 0.00048
#> • N discovery pairs called as significant: 37/1911
# 8. write results to a directory. tempdir() is used so this example is
# self-contained; for a real analysis choose a directory you can find again.
output_dir <- file.path(tempdir(), "sceptre_outputs_lowmoi")
write_outputs_to_directory(
sceptre_object = sceptre_object,
directory = output_dir
)
message(
"sceptre outputs written to a temporary directory; ",
'open with `browseURL("', output_dir, '")`'
)
#> sceptre outputs written to a temporary directory; open with `browseURL("/var/folders/1w/h831hyps5qs5lzkh5xjj0_wh0000gq/T//RtmpgcetiN/sceptre_outputs_lowmoi")`
##########################
# High-MOI CRISPRi example
##########################
# 1. create the sceptre object from cellranger output
directories <- paste0(
system.file("extdata", package = "sceptre"),
"/highmoi_example/gem_group_", c(1, 2)
)
data(grna_target_data_frame_highmoi)
sceptre_object <- import_data_from_cellranger(
directories = directories,
moi = "high",
grna_target_data_frame = grna_target_data_frame_highmoi
)
#> Processing directory 1.
#> ✓
#> Processing directory 2.
#> ✓
#> Combining matrices across directories.
#> ✓
#> Creating the sceptre object.
#> ✓
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 500 cells
#> • 100 responses
#> • High multiplicity-of-infection
#> • 50 targeting gRNAs (distributed across 25 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✗ set_analysis_parameters()
#> ✗ assign_grnas()
#> ✗ run_qc()
#> ✗ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: not specified
#> • Positive control pairs: not specified
#> • Sidedness of test: not specified
#> • Resampling mechanism: not specified
#> • gRNA integration strategy: not specified
#> • Resampling approximation: not specified
#> • Multiple testing adjustment: none
#> • N nonzero treatment cells threshold: not specified
#> • N nonzero control cells threshold: not specified
#> • Formula object: not specified
# 2. set the analysis parameters
positive_control_pairs <- construct_positive_control_pairs(sceptre_object)
discovery_pairs <- construct_cis_pairs(sceptre_object,
positive_control_pairs = positive_control_pairs,
distance_threshold = 5e6
)
sceptre_object <- set_analysis_parameters(
sceptre_object = sceptre_object,
discovery_pairs = discovery_pairs,
positive_control_pairs = positive_control_pairs,
side = "left"
)
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 500 cells
#> • 100 responses
#> • High multiplicity-of-infection
#> • 50 targeting gRNAs (distributed across 25 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✗ assign_grnas()
#> ✗ run_qc()
#> ✗ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1406 pairs
#> • Positive control pairs: data frame with 5 pairs
#> • Sidedness of test: left
#> • Resampling mechanism: conditional resampling
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: not specified
#> • N nonzero control cells threshold: not specified
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
# 3. assign grnas
plot_grna_count_distributions(sceptre_object)
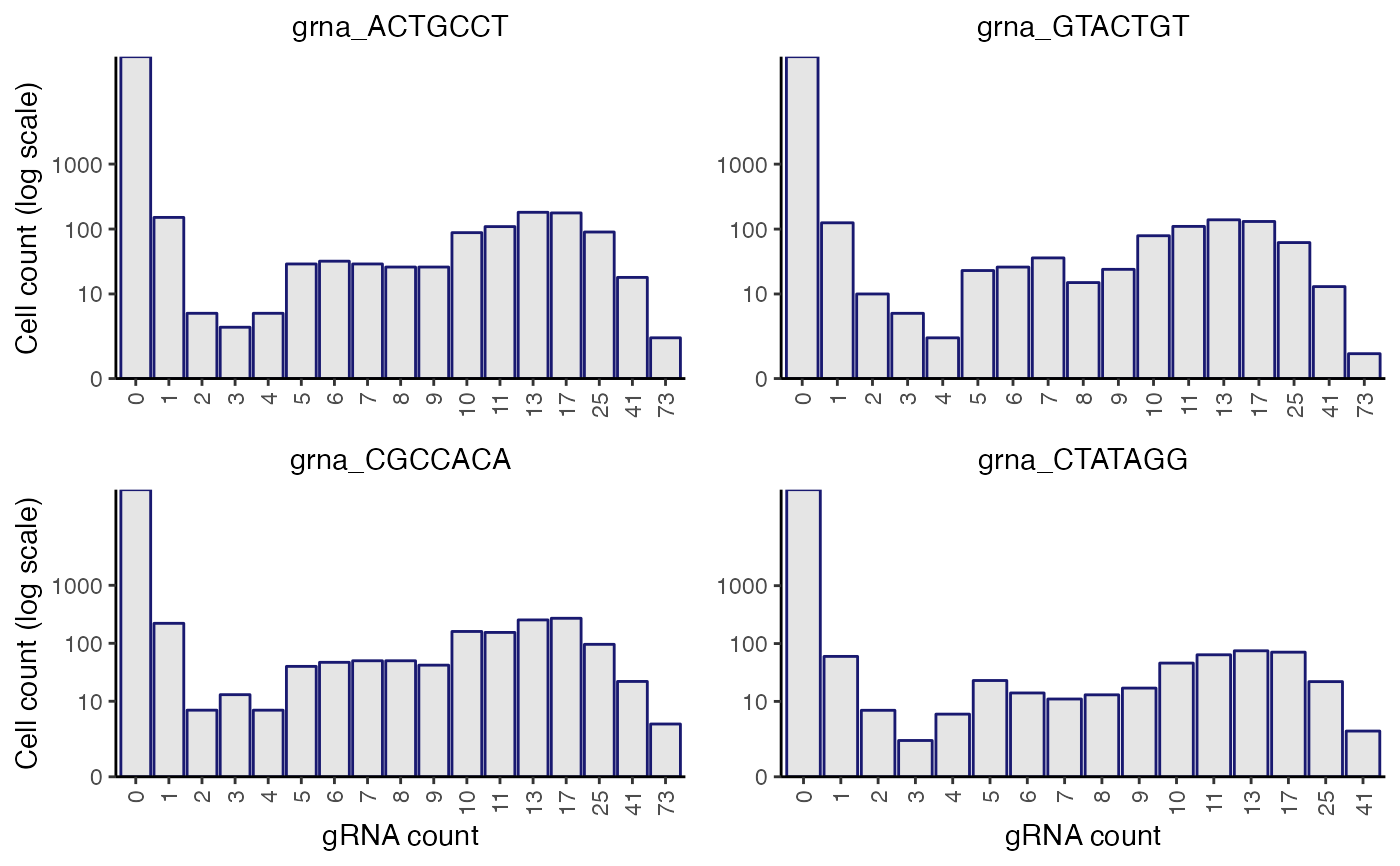 sceptre_object <- sceptre_object |> assign_grnas()
#> Note: If you are on a Mac laptop or desktop, consider setting `parallel = TRUE` to improve speed. Otherwise, keep `parallel = FALSE`.
#> Performing gRNA-to-cell assignments for gRNA ENSG00000224277_grna1 (1 of 60)
#> Performing gRNA-to-cell assignments for gRNA ENSG00000226772_grna1 (5 of 60)
#> Performing gRNA-to-cell assignments for gRNA ENSG00000286326_grna2 (10 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_3_grna1 (15 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_5_grna2 (20 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_8_grna1 (25 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_10_grna2 (30 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_13_grna1 (35 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_15_grna2 (40 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_18_grna1 (45 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_20_grna2 (50 of 60)
#> Performing gRNA-to-cell assignments for gRNA non-targeting_grna5 (55 of 60)
#> Performing gRNA-to-cell assignments for gRNA non-targeting_grna10 (60 of 60)
plot(sceptre_object)
sceptre_object <- sceptre_object |> assign_grnas()
#> Note: If you are on a Mac laptop or desktop, consider setting `parallel = TRUE` to improve speed. Otherwise, keep `parallel = FALSE`.
#> Performing gRNA-to-cell assignments for gRNA ENSG00000224277_grna1 (1 of 60)
#> Performing gRNA-to-cell assignments for gRNA ENSG00000226772_grna1 (5 of 60)
#> Performing gRNA-to-cell assignments for gRNA ENSG00000286326_grna2 (10 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_3_grna1 (15 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_5_grna2 (20 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_8_grna1 (25 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_10_grna2 (30 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_13_grna1 (35 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_15_grna2 (40 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_18_grna1 (45 of 60)
#> Performing gRNA-to-cell assignments for gRNA candidate_enh_20_grna2 (50 of 60)
#> Performing gRNA-to-cell assignments for gRNA non-targeting_grna5 (55 of 60)
#> Performing gRNA-to-cell assignments for gRNA non-targeting_grna10 (60 of 60)
plot(sceptre_object)
 print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 500 cells
#> • 100 responses
#> • High multiplicity-of-infection
#> • 50 targeting gRNAs (distributed across 25 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✗ run_qc()
#> ✗ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1406 pairs
#> • Positive control pairs: data frame with 5 pairs
#> • Sidedness of test: left
#> • Resampling mechanism: conditional resampling
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: not specified
#> • N nonzero control cells threshold: not specified
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: mixture
#> • Mean N cells per gRNA: 58.35
#> • Mean N gRNAs per cell (MOI): 7
#> • gRNA assignment formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
# 4. run qc
plot_covariates(sceptre_object, p_mito_threshold = 0.075)
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 500 cells
#> • 100 responses
#> • High multiplicity-of-infection
#> • 50 targeting gRNAs (distributed across 25 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✗ run_qc()
#> ✗ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1406 pairs
#> • Positive control pairs: data frame with 5 pairs
#> • Sidedness of test: left
#> • Resampling mechanism: conditional resampling
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: not specified
#> • N nonzero control cells threshold: not specified
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: mixture
#> • Mean N cells per gRNA: 58.35
#> • Mean N gRNAs per cell (MOI): 7
#> • gRNA assignment formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
# 4. run qc
plot_covariates(sceptre_object, p_mito_threshold = 0.075)
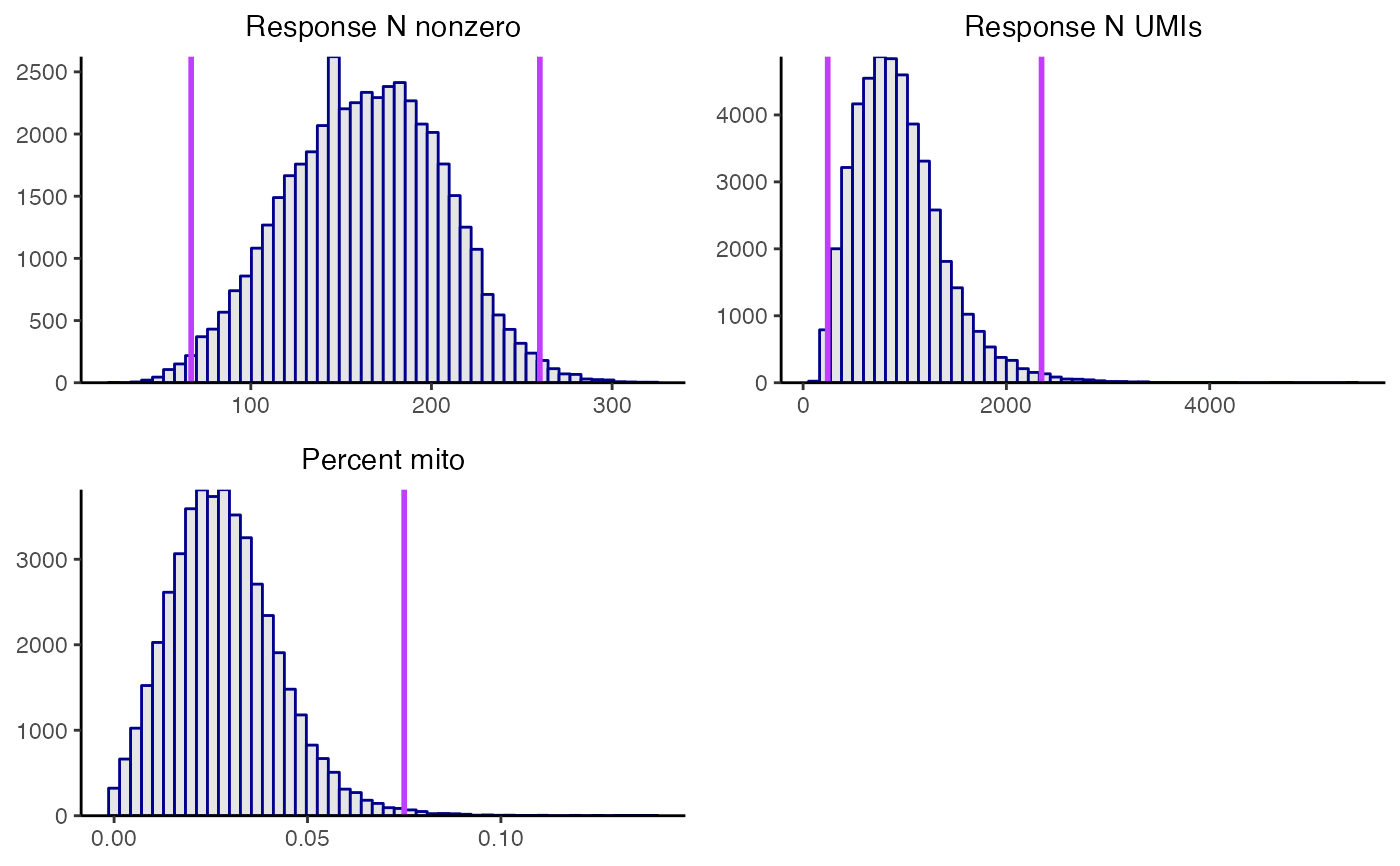 sceptre_object <- sceptre_object |> run_qc(p_mito_threshold = 0.075)
plot(sceptre_object)
sceptre_object <- sceptre_object |> run_qc(p_mito_threshold = 0.075)
plot(sceptre_object)
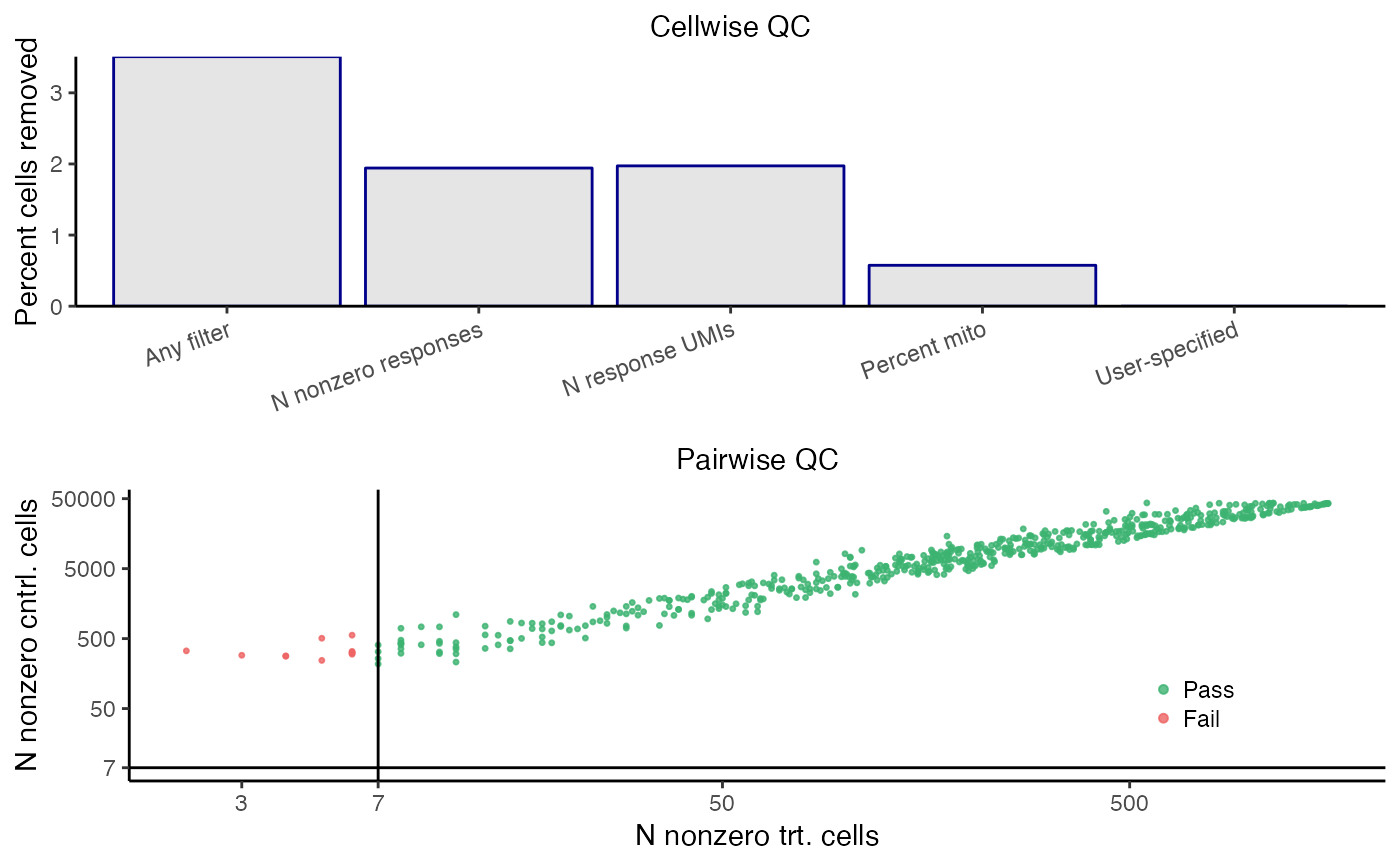 print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 500 cells (483 after cellwise QC)
#> • 100 responses
#> • High multiplicity-of-infection
#> • 50 targeting gRNAs (distributed across 25 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✗ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1406 pairs (1314 after pairwise QC)
#> • Positive control pairs: data frame with 5 pairs (5 after pairwise QC)
#> • Sidedness of test: left
#> • Resampling mechanism: conditional resampling
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: mixture
#> • Mean N cells per gRNA: 58.35
#> • Mean N gRNAs per cell (MOI): 7
#> • gRNA assignment formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
# 5. run the calibration check
sceptre_object <- run_calibration_check(sceptre_object)
#> Note: If you are on a Mac laptop or desktop, consider setting `parallel = TRUE` to improve speed. Otherwise, keep `parallel = FALSE`.
#> Constructing negative control pairs.
#> ✓
#> Running precomputation on response ENSG00000253631 (1 of 98)
#> Running precomputation on response ENSG00000100053 (5 of 98)
#> Running precomputation on response ENSG00000100325 (10 of 98)
#> Running precomputation on response ENSG00000253963 (15 of 98)
#> Running precomputation on response ENSG00000100314 (20 of 98)
#> Running precomputation on response ENSG00000273343 (25 of 98)
#> Running precomputation on response ENSG00000133475 (30 of 98)
#> Running precomputation on response ENSG00000234503 (35 of 98)
#> Running precomputation on response ENSG00000253126 (40 of 98)
#> Running precomputation on response ENSG00000235786 (45 of 98)
#> Running precomputation on response ENSG00000183765 (50 of 98)
#> Running precomputation on response ENSG00000161133 (55 of 98)
#> Running precomputation on response ENSG00000234208 (60 of 98)
#> Running precomputation on response ENSG00000211661 (65 of 98)
#> Running precomputation on response ENSG00000211672 (70 of 98)
#> Running precomputation on response ENSG00000220891 (75 of 98)
#> Running precomputation on response ENSG00000227838 (80 of 98)
#> Running precomputation on response ENSG00000128271 (85 of 98)
#> Running precomputation on response ENSG00000099958 (90 of 98)
#> Running precomputation on response ENSG00000167037 (95 of 98)
#> Analyzing pairs containing gRNA group non-targeting_grna1&non-targeting_grna6 (1 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna3&non-targeting_grna5 (5 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna9&non-targeting_grna10 (10 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna8&non-targeting_grna10 (15 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna1&non-targeting_grna5 (20 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna7&non-targeting_grna10 (25 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna2&non-targeting_grna9 (30 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna2&non-targeting_grna4 (35 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna2&non-targeting_grna5 (40 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna1&non-targeting_grna2 (45 of 45)
plot(sceptre_object)
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 500 cells (483 after cellwise QC)
#> • 100 responses
#> • High multiplicity-of-infection
#> • 50 targeting gRNAs (distributed across 25 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✗ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1406 pairs (1314 after pairwise QC)
#> • Positive control pairs: data frame with 5 pairs (5 after pairwise QC)
#> • Sidedness of test: left
#> • Resampling mechanism: conditional resampling
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: mixture
#> • Mean N cells per gRNA: 58.35
#> • Mean N gRNAs per cell (MOI): 7
#> • gRNA assignment formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
# 5. run the calibration check
sceptre_object <- run_calibration_check(sceptre_object)
#> Note: If you are on a Mac laptop or desktop, consider setting `parallel = TRUE` to improve speed. Otherwise, keep `parallel = FALSE`.
#> Constructing negative control pairs.
#> ✓
#> Running precomputation on response ENSG00000253631 (1 of 98)
#> Running precomputation on response ENSG00000100053 (5 of 98)
#> Running precomputation on response ENSG00000100325 (10 of 98)
#> Running precomputation on response ENSG00000253963 (15 of 98)
#> Running precomputation on response ENSG00000100314 (20 of 98)
#> Running precomputation on response ENSG00000273343 (25 of 98)
#> Running precomputation on response ENSG00000133475 (30 of 98)
#> Running precomputation on response ENSG00000234503 (35 of 98)
#> Running precomputation on response ENSG00000253126 (40 of 98)
#> Running precomputation on response ENSG00000235786 (45 of 98)
#> Running precomputation on response ENSG00000183765 (50 of 98)
#> Running precomputation on response ENSG00000161133 (55 of 98)
#> Running precomputation on response ENSG00000234208 (60 of 98)
#> Running precomputation on response ENSG00000211661 (65 of 98)
#> Running precomputation on response ENSG00000211672 (70 of 98)
#> Running precomputation on response ENSG00000220891 (75 of 98)
#> Running precomputation on response ENSG00000227838 (80 of 98)
#> Running precomputation on response ENSG00000128271 (85 of 98)
#> Running precomputation on response ENSG00000099958 (90 of 98)
#> Running precomputation on response ENSG00000167037 (95 of 98)
#> Analyzing pairs containing gRNA group non-targeting_grna1&non-targeting_grna6 (1 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna3&non-targeting_grna5 (5 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna9&non-targeting_grna10 (10 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna8&non-targeting_grna10 (15 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna1&non-targeting_grna5 (20 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna7&non-targeting_grna10 (25 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna2&non-targeting_grna9 (30 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna2&non-targeting_grna4 (35 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna2&non-targeting_grna5 (40 of 45)
#> Analyzing pairs containing gRNA group non-targeting_grna1&non-targeting_grna2 (45 of 45)
plot(sceptre_object)
 print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 500 cells (483 after cellwise QC)
#> • 100 responses
#> • High multiplicity-of-infection
#> • 50 targeting gRNAs (distributed across 25 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✓ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1406 pairs (1314 after pairwise QC)
#> • Positive control pairs: data frame with 5 pairs (5 after pairwise QC)
#> • Sidedness of test: left
#> • Resampling mechanism: conditional resampling
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: mixture
#> • Mean N cells per gRNA: 58.35
#> • Mean N gRNAs per cell (MOI): 7
#> • gRNA assignment formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> Summary of results:
#> • N negative control pairs called as significant: 0/1314
#> • Mean log-2 FC for negative control pairs: 0.002
# 6. run the power check
sceptre_object <- run_power_check(sceptre_object)
#> Note: If you are on a Mac laptop or desktop, consider setting `parallel = TRUE` to improve speed. Otherwise, keep `parallel = FALSE`.
#> Running precomputation on response ENSG00000224277 (1 of 5)
#> Running precomputation on response ENSG00000226772 (5 of 5)
#> Analyzing pairs containing gRNA group ENSG00000224277 (1 of 5)
#> Analyzing pairs containing gRNA group ENSG00000226772 (5 of 5)
plot(sceptre_object)
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 500 cells (483 after cellwise QC)
#> • 100 responses
#> • High multiplicity-of-infection
#> • 50 targeting gRNAs (distributed across 25 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✓ run_calibration_check()
#> ✗ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1406 pairs (1314 after pairwise QC)
#> • Positive control pairs: data frame with 5 pairs (5 after pairwise QC)
#> • Sidedness of test: left
#> • Resampling mechanism: conditional resampling
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: mixture
#> • Mean N cells per gRNA: 58.35
#> • Mean N gRNAs per cell (MOI): 7
#> • gRNA assignment formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> Summary of results:
#> • N negative control pairs called as significant: 0/1314
#> • Mean log-2 FC for negative control pairs: 0.002
# 6. run the power check
sceptre_object <- run_power_check(sceptre_object)
#> Note: If you are on a Mac laptop or desktop, consider setting `parallel = TRUE` to improve speed. Otherwise, keep `parallel = FALSE`.
#> Running precomputation on response ENSG00000224277 (1 of 5)
#> Running precomputation on response ENSG00000226772 (5 of 5)
#> Analyzing pairs containing gRNA group ENSG00000224277 (1 of 5)
#> Analyzing pairs containing gRNA group ENSG00000226772 (5 of 5)
plot(sceptre_object)
 print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 500 cells (483 after cellwise QC)
#> • 100 responses
#> • High multiplicity-of-infection
#> • 50 targeting gRNAs (distributed across 25 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✓ run_calibration_check()
#> ✓ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1406 pairs (1314 after pairwise QC)
#> • Positive control pairs: data frame with 5 pairs (5 after pairwise QC)
#> • Sidedness of test: left
#> • Resampling mechanism: conditional resampling
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: mixture
#> • Mean N cells per gRNA: 58.35
#> • Mean N gRNAs per cell (MOI): 7
#> • gRNA assignment formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> Summary of results:
#> • N negative control pairs called as significant: 0/1314
#> • Mean log-2 FC for negative control pairs: 0.002
#> • Median positive control p-value: 2.2e-10
# 7. run discovery analysis
sceptre_object <- run_discovery_analysis(sceptre_object)
#> Note: If you are on a Mac laptop or desktop, consider setting `parallel = TRUE` to improve speed. Otherwise, keep `parallel = FALSE`.
#> Running precomputation on response ENSG00000100218 (1 of 97)
#> Running precomputation on response ENSG00000099958 (5 of 97)
#> Running precomputation on response ENSG00000211638 (10 of 97)
#> Running precomputation on response ENSG00000211685 (15 of 97)
#> Running precomputation on response ENSG00000220891 (20 of 97)
#> Running precomputation on response ENSG00000187905 (25 of 97)
#> Running precomputation on response ENSG00000235954 (30 of 97)
#> Running precomputation on response ENSG00000244486 (35 of 97)
#> Running precomputation on response ENSG00000225783 (40 of 97)
#> Running precomputation on response ENSG00000224277 (45 of 97)
#> Running precomputation on response ENSG00000286326 (50 of 97)
#> Running precomputation on response ENSG00000211672 (55 of 97)
#> Running precomputation on response ENSG00000203280 (60 of 97)
#> Running precomputation on response ENSG00000233521 (65 of 97)
#> Running precomputation on response ENSG00000100319 (70 of 97)
#> Running precomputation on response ENSG00000211674 (75 of 97)
#> Running precomputation on response ENSG00000099917 (80 of 97)
#> Running precomputation on response ENSG00000100053 (85 of 97)
#> Running precomputation on response ENSG00000229770 (90 of 97)
#> Running precomputation on response ENSG00000253920 (95 of 97)
#> Analyzing pairs containing gRNA group candidate_enh_1 (1 of 20)
#> Analyzing pairs containing gRNA group candidate_enh_5 (5 of 20)
#> Analyzing pairs containing gRNA group candidate_enh_12 (10 of 20)
#> Analyzing pairs containing gRNA group candidate_enh_19 (15 of 20)
#> Analyzing pairs containing gRNA group candidate_enh_14 (20 of 20)
plot(sceptre_object)
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 500 cells (483 after cellwise QC)
#> • 100 responses
#> • High multiplicity-of-infection
#> • 50 targeting gRNAs (distributed across 25 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✓ run_calibration_check()
#> ✓ run_power_check()
#> ✗ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1406 pairs (1314 after pairwise QC)
#> • Positive control pairs: data frame with 5 pairs (5 after pairwise QC)
#> • Sidedness of test: left
#> • Resampling mechanism: conditional resampling
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: mixture
#> • Mean N cells per gRNA: 58.35
#> • Mean N gRNAs per cell (MOI): 7
#> • gRNA assignment formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> Summary of results:
#> • N negative control pairs called as significant: 0/1314
#> • Mean log-2 FC for negative control pairs: 0.002
#> • Median positive control p-value: 2.2e-10
# 7. run discovery analysis
sceptre_object <- run_discovery_analysis(sceptre_object)
#> Note: If you are on a Mac laptop or desktop, consider setting `parallel = TRUE` to improve speed. Otherwise, keep `parallel = FALSE`.
#> Running precomputation on response ENSG00000100218 (1 of 97)
#> Running precomputation on response ENSG00000099958 (5 of 97)
#> Running precomputation on response ENSG00000211638 (10 of 97)
#> Running precomputation on response ENSG00000211685 (15 of 97)
#> Running precomputation on response ENSG00000220891 (20 of 97)
#> Running precomputation on response ENSG00000187905 (25 of 97)
#> Running precomputation on response ENSG00000235954 (30 of 97)
#> Running precomputation on response ENSG00000244486 (35 of 97)
#> Running precomputation on response ENSG00000225783 (40 of 97)
#> Running precomputation on response ENSG00000224277 (45 of 97)
#> Running precomputation on response ENSG00000286326 (50 of 97)
#> Running precomputation on response ENSG00000211672 (55 of 97)
#> Running precomputation on response ENSG00000203280 (60 of 97)
#> Running precomputation on response ENSG00000233521 (65 of 97)
#> Running precomputation on response ENSG00000100319 (70 of 97)
#> Running precomputation on response ENSG00000211674 (75 of 97)
#> Running precomputation on response ENSG00000099917 (80 of 97)
#> Running precomputation on response ENSG00000100053 (85 of 97)
#> Running precomputation on response ENSG00000229770 (90 of 97)
#> Running precomputation on response ENSG00000253920 (95 of 97)
#> Analyzing pairs containing gRNA group candidate_enh_1 (1 of 20)
#> Analyzing pairs containing gRNA group candidate_enh_5 (5 of 20)
#> Analyzing pairs containing gRNA group candidate_enh_12 (10 of 20)
#> Analyzing pairs containing gRNA group candidate_enh_19 (15 of 20)
#> Analyzing pairs containing gRNA group candidate_enh_14 (20 of 20)
plot(sceptre_object)
 print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 500 cells (483 after cellwise QC)
#> • 100 responses
#> • High multiplicity-of-infection
#> • 50 targeting gRNAs (distributed across 25 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✓ run_calibration_check()
#> ✓ run_power_check()
#> ✓ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1406 pairs (1314 after pairwise QC)
#> • Positive control pairs: data frame with 5 pairs (5 after pairwise QC)
#> • Sidedness of test: left
#> • Resampling mechanism: conditional resampling
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: mixture
#> • Mean N cells per gRNA: 58.35
#> • Mean N gRNAs per cell (MOI): 7
#> • gRNA assignment formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> Summary of results:
#> • N negative control pairs called as significant: 0/1314
#> • Mean log-2 FC for negative control pairs: 0.002
#> • Median positive control p-value: 2.2e-10
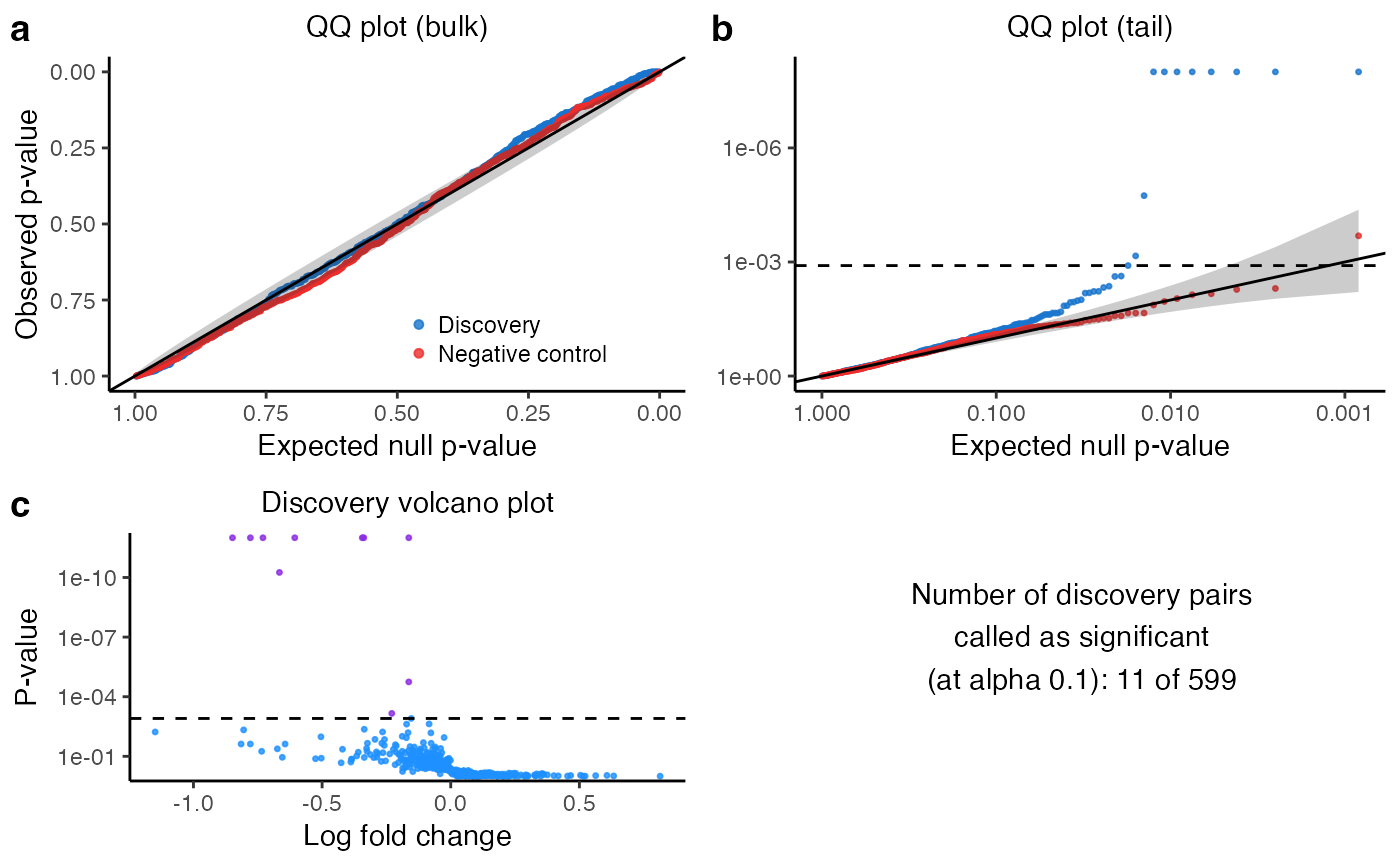
#> • N discovery pairs called as significant: 10/1314
# 8. write results to a directory. tempdir() is used so this example is
# self-contained; for a real analysis choose a directory you can find again.
output_dir <- file.path(tempdir(), "sceptre_outputs_highmoi")
write_outputs_to_directory(
sceptre_object = sceptre_object,
directory = output_dir
)
message(
"sceptre outputs written to a temporary directory; ",
'open by running `browseURL("', output_dir, '")` in the console.'
)
#> sceptre outputs written to a temporary directory; open by running `browseURL("/var/folders/1w/h831hyps5qs5lzkh5xjj0_wh0000gq/T//RtmpgcetiN/sceptre_outputs_highmoi")` in the console.
print(sceptre_object)
#> An object of class sceptre_object.
#>
#> Attributes of the data:
#> • 500 cells (483 after cellwise QC)
#> • 100 responses
#> • High multiplicity-of-infection
#> • 50 targeting gRNAs (distributed across 25 targets)
#> • 10 non-targeting gRNAs
#> • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)
#>
#> Analysis status:
#> ✓ import_data()
#> ✓ set_analysis_parameters()
#> ✓ assign_grnas()
#> ✓ run_qc()
#> ✓ run_calibration_check()
#> ✓ run_power_check()
#> ✓ run_discovery_analysis()
#>
#> Analysis parameters:
#> • Discovery pairs: data frame with 1406 pairs (1314 after pairwise QC)
#> • Positive control pairs: data frame with 5 pairs (5 after pairwise QC)
#> • Sidedness of test: left
#> • Resampling mechanism: conditional resampling
#> • gRNA integration strategy: union
#> • Resampling approximation: skew normal
#> • Multiple testing adjustment: BH at level 0.1
#> • N nonzero treatment cells threshold: 7
#> • N nonzero control cells threshold: 7
#> • Formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> gRNA-to-cell assignment information:
#> • Assignment method: mixture
#> • Mean N cells per gRNA: 58.35
#> • Mean N gRNAs per cell (MOI): 7
#> • gRNA assignment formula object: log(response_n_nonzero) + log(response_n_umis) + log(grna_n_nonzero) + log(grna_n_umis) + batch
#>
#> Summary of results:
#> • N negative control pairs called as significant: 0/1314
#> • Mean log-2 FC for negative control pairs: 0.002
#> • Median positive control p-value: 2.2e-10
#> • N discovery pairs called as significant: 10/1314
# 8. write results to a directory. tempdir() is used so this example is
# self-contained; for a real analysis choose a directory you can find again.
output_dir <- file.path(tempdir(), "sceptre_outputs_highmoi")
write_outputs_to_directory(
sceptre_object = sceptre_object,
directory = output_dir
)
message(
"sceptre outputs written to a temporary directory; ",
'open by running `browseURL("', output_dir, '")` in the console.'
)
#> sceptre outputs written to a temporary directory; open by running `browseURL("/var/folders/1w/h831hyps5qs5lzkh5xjj0_wh0000gq/T//RtmpgcetiN/sceptre_outputs_highmoi")` in the console.