sceptre is an R package that facilitates statistically
rigorous, massively scalable, and user-friendly single-cell CRISPR
screen data analysis. To get started, users should install
sceptre.
install.packages("devtools")
devtools::install_github("katsevich-lab/sceptre")See the frequently
asked questions page for tips on installing sceptre
such that it runs as fast as possible. Users can load
sceptre via a call to library().
The standard pipeline involved in applying sceptre to
analyze a dataset consists of several steps, which we summarize in the
following schematic.
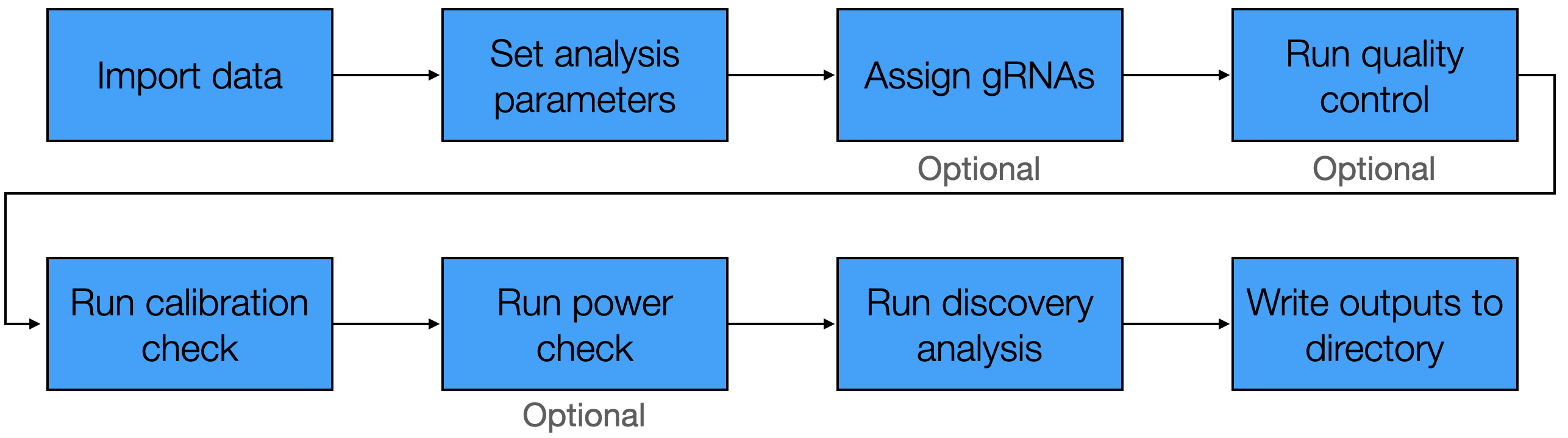
Standard sceptre pipeline
This chapter illustrates application of sceptre to a
small simulated CRISPRi screen of candidate enhancers, modeled on that
of Gasperini,
2019. The goal of the analysis is to confidently link enhancers to
genes by testing for changes in gene expression in response to the
CRISPR perturbations of the candidate enhancers.
Using sceptre is simple; carrying out an entire analysis
requires only a few lines of code. Below, we provide a minimal working
example of applying sceptre to analyze the example data.
First, we load the example data into R via the function
import_data_from_cellranger(), which creates a
sceptre_object, an object-based representation of the
single-cell CRISPR screen data. Next, we specify the gRNA-gene pairs
that we seek to test for association. Then, we call the pipeline
functions on the sceptre_object in order. Finally, we write
the outputs to a temporary directory.
# load the data, creating a sceptre_object
directories <- paste0(
system.file("extdata", package = "sceptre"),
"/highmoi_example/gem_group_",
1:2
)
data(grna_target_data_frame_highmoi)
sceptre_object <- import_data_from_cellranger(
directories = directories,
moi = "high",
grna_target_data_frame = grna_target_data_frame_highmoi
)
# construct the grna-gene pairs to analyze
positive_control_pairs <- construct_positive_control_pairs(sceptre_object)
discovery_pairs <- construct_cis_pairs(
sceptre_object,
positive_control_pairs = positive_control_pairs
)
# apply the pipeline functions to the sceptre_object in order
sceptre_object <- sceptre_object |> # |> is R's base pipe, similar to %>%
set_analysis_parameters(discovery_pairs, positive_control_pairs) |>
run_calibration_check() |>
run_power_check() |>
run_discovery_analysis()
# write the results to disk
output_directory <- file.path(tempdir(), "sceptre_outputs")
write_outputs_to_directory(sceptre_object, output_directory)
# open the output directory to inspect the results
browseURL(output_directory)That’s it. The output directory now contains a variety of results and plots from the analysis, including the set of significant gRNA-gene pairs:
## # A tibble: 13 × 4
## grna_target response_id p_value log_2_fold_change
## <chr> <chr> <dbl> <dbl>
## 1 candidate_enh_17 ENSG00000220891 0.00000215 -1.50
## 2 candidate_enh_15 ENSG00000211641 0.00000604 -0.743
## 3 candidate_enh_18 ENSG00000220891 0.0000262 -1.29
## 4 candidate_enh_5 ENSG00000211655 0.0000310 -1.11
## 5 candidate_enh_19 ENSG00000253451 0.0000865 -0.874
## # ℹ 8 more rowsWe describe each step of the pipeline in greater detail below.
1. Import data
The first step is to import the data. Data can be imported
into sceptre from 10x Cell Ranger or Parse outputs, as well
as from R matrices. The simplest way to import the data is to
read the output of one or more calls to cellranger_count
into sceptre via the function
import_data_from_cellranger().
import_data_from_cellranger() requires three arguments:
directories, grna_target_data_frame, and
moi.
-
directoriesis a character vector specifying the locations of the directories outputted by one or more calls tocellranger_count. Below, we set the variabledirectoriesto the (machine-dependent) location of the example CRISPRi data on disk.directories <- paste0( system.file("extdata", package = "sceptre"), "/highmoi_example/gem_group_", 1:2 ) directories # file paths to the example data on your computer## [1] "/private/var/folders/1w/h831hyps5qs5lzkh5xjj0_wh0000gq/T/RtmpgcetiN/temp_libpath124931c84cecb/sceptre/extdata/highmoi_example/gem_group_1" ## [2] "/private/var/folders/1w/h831hyps5qs5lzkh5xjj0_wh0000gq/T/RtmpgcetiN/temp_libpath124931c84cecb/sceptre/extdata/highmoi_example/gem_group_2"directoriespoints to two directories, both of which store the expression data in matrix market format and contain the filesbarcodes.tsv.gz,features.tsv.gz, andmatrix.mtx.gz.list.files(directories[1])## [1] "barcodes.tsv.gz" "features.tsv.gz" "matrix.mtx.gz"list.files(directories[2])## [1] "barcodes.tsv.gz" "features.tsv.gz" "matrix.mtx.gz" -
grna_target_data_frameis a data frame mapping each individual gRNA to the genomic element that the gRNA targets.grna_target_data_framecontains two required columns:grna_idandgrna_target.grna_idis the ID of an individual gRNA, whilegrna_targetis a label specifying the genomic element that the gRNA targets. (Typically, multiple gRNAs are designed to target a given genomic element in a single-cell CRISPR screen.) Non-targeting (NT) gRNAs are assigned a gRNA target label of “non-targeting”.grna_target_data_frameoptionally contains the columnschr,start, andend, which give the chromosome, start coordinate, and end coordinate, respectively, of the genomic region that each gRNA targets. Finally,grna_target_data_frameoptionally can contain the columnvector_idspecifying the vector to which a given gRNA belongs.vector_idshould be supplied in experiments in which each viral vector contains two or more distinct gRNAs (as in, e.g., (Replogle, 2022)). We load and examine thegrna_target_data_framecorresponding to the example data.## grna_id grna_target chr start end ## 1 ENSG00000224277_grna1 ENSG00000224277 chr22 23567064 23567113 ## 2 ENSG00000224277_grna2 ENSG00000224277 chr22 23567114 23567163 ## 3 ENSG00000233521_grna1 ENSG00000233521 chr22 27225134 27225183 ## 4 ENSG00000233521_grna2 ENSG00000233521 chr22 27225184 27225233 ## 11 candidate_enh_1_grna1 candidate_enh_1 chr22 20772896 20772945 ## 12 candidate_enh_1_grna2 candidate_enh_1 chr22 20772946 20772995 ## 13 candidate_enh_2_grna1 candidate_enh_2 chr22 19998415 19998464 ## 14 candidate_enh_2_grna2 candidate_enh_2 chr22 19998465 19998514 ## 51 non-targeting_grna1 non-targeting <NA> NA NA ## 52 non-targeting_grna2 non-targeting <NA> NA NA ## 53 non-targeting_grna3 non-targeting <NA> NA NA ## 54 non-targeting_grna4 non-targeting <NA> NA NASome gRNAs (e.g.,
ENSG00000224277_grna1) target gene transcription start sites and serve as positive controls; other gRNAs (e.g.,candidate_enh_1_grna1) target candidate enhancers, while others still (e.g.,non-targeting_grna1) are non-targeting. Each gene and candidate enhancer in this dataset is targeted by exactly two gRNAs. -
moiis a string specifying the multiplicity-of-infection (MOI) of the data, taking values"high"or"low". A high-MOI (respectively, low-MOI) dataset is one in which the experimenter has aimed to insert multiple gRNAs (respectively, a single gRNA) into each cell. (If a given cell is determined to contain multiple gRNAs in a low-MOI screen, that cell is removed as part of the quality control step, as discussed below.) The example dataset is a high MOI dataset, and so we setmoito"high".moi <- "high"
Finally, we call the function
import_data_from_cellranger(), passing
directories, grna_target_data_frame, and
moi as arguments.
sceptre_object <- import_data_from_cellranger(
directories = directories,
grna_target_data_frame = grna_target_data_frame_highmoi,
moi = moi
)import_data_from_cellranger() returns a
sceptre_object, which is an object-based representation of
the single-cell CRISPR screen data. Evaluating
sceptre_object in the console prints a helpful summary of
the data.
sceptre_object## An object of class sceptre_object.
##
## Attributes of the data:
## • 500 cells
## • 100 responses
## • High multiplicity-of-infection
## • 50 targeting gRNAs (distributed across 25 targets)
## • 10 non-targeting gRNAs
## • 5 covariates (batch, grna_n_nonzero, grna_n_umis, response_n_nonzero, response_n_umis)Several metrics are displayed, including the number of cells, the
number of genes (or “responses”), and the number of gRNAs present in the
data. sceptre also automatically computes the following
cell-specific covariates: grna_n_nonzero (i.e., the number
of gRNAs expressed in the cell), grna_n_umis (i.e., the
number of gRNA UMIs sequenced in the cell),
response_n_nonzero (i.e., the number of responses expressed
in the cell), response_n_umis (i.e., the number of response
UMIs sequenced in the cell), response_p_mito (i.e., the
fraction of transcripts mapping to mitochondrial genes), and
batch. (Cells loaded from different directories are assumed
to come from different batches.)
2. Set analysis parameters
The second step is to set the analysis parameters. The most important analysis parameters are the discovery pairs, positive control pairs, sidedness, and gRNA grouping strategy.
-
Discovery pairs and positive control pairs. The primary goal of
sceptreis to determine whether perturbation of a gRNA target (such as an enhancer) leads to a change in expression of a response (such as gene). We use the term target-response pair to refer to a given gRNA target and response that we seek to test for association (upon perturbation of the gRNA target). A discovery target-response pair is a target-response pair whose association status we do not know but would like to learn. For example, in an experiment in which we aim to link putative enhancers to genes, the discovery target-response pairs might consist of the set of putative enhancers and genes in close physical proximity to one another.A positive control (resp., negative control) target-response pair is a target-response pair for which we know that there is (resp., is not) a relationship between the target and the response. Positive control target-response pairs often are formed by coupling a transcription start site to the gene known to be regulated by that transcription start site. Negative control target-response pairs, meanwhile, typically are constructed by pairing negative control gRNAs to one or more responses. (We defer a detailed discussion of negative control pairs to a later section of this chapter.) Discovery pairs are of primary scientific interest, while positive control and negative control pairs serve a mainly technial purpose, helping us verify that the biological assay and statistical methodology are in working order.
sceptreoffers several helper functions to facilitate the construction of positive control and discovery pairs. The functionconstruct_positive_control_pairs()takes as argument asceptre_objectand outputs the set of positive control pairs formed by matching gRNA targets (as contained in thegrna_target_data_frame) to response IDs. Positive control pairs are optional and need not be computed.positive_control_pairs <- construct_positive_control_pairs(sceptre_object) head(positive_control_pairs)## grna_target response_id ## 1 ENSG00000224277 ENSG00000224277 ## 2 ENSG00000233521 ENSG00000233521 ## 3 ENSG00000226772 ENSG00000226772 ## 4 ENSG00000234503 ENSG00000234503 ## 5 ENSG00000286326 ENSG00000286326Next, the functions
construct_cis_pairs()andconstruct_trans_pairs()facilitate the construction of cis and trans discovery sets, respectively.construct_cis_pairs()takes as arguments asceptre_objectand an integerdistance_thresholdand returns the set of response-target pairs located on the same chromosome withindistance_thresholdbases of one another.positive_control_pairsoptionally can be passed to this function, in which case positive control gRNA targets are excluded from the cis pairs. (Note thatconstruct_cis_pairs()assumes that the responses are genes rather than, say, proteins or chromatin-derived features.)discovery_pairs <- construct_cis_pairs( sceptre_object = sceptre_object, positive_control_pairs = positive_control_pairs, distance_threshold = 5e6 ) discovery_pairs[c(1:4, 101:104), ]## grna_target response_id ## 1 candidate_enh_1 ENSG00000099889 ## 2 candidate_enh_1 ENSG00000040608 ## 3 candidate_enh_1 ENSG00000273343 ## 4 candidate_enh_1 ENSG00000161133 ## 101 candidate_enh_2 ENSG00000211638 ## 102 candidate_enh_2 ENSG00000211640 ## 103 candidate_enh_2 ENSG00000253126 ## 104 candidate_enh_2 ENSG00000211641construct_trans_pairs()constructs the entire set of possible target-response pairs. -
Sidedness. The parameter
sidecontrols whether to run a left-tailed ("left"), right-tailed ("right"), or two-tailed ("both"; default) test. A left-tailed (resp., right-tailed) test is appropriate when testing for a decrease (resp., increase) in expression; a two-tailed test, by contrast, is appropriate when testing for an increase or decrease in expression. A left-tailed test is the most appropriate choice for a CRISPRi screen of enhancers, and so we setsideto"left".side <- "left" gRNA integration strategy. Typically, multiple gRNAs are designed to target a given genomic element. The parameter
grna_integration_strategycontrols if and how gRNAs that target the same genomic element are integrated. The default option,"union", combines gRNAs that target the same element into a single “grouped gRNA;” this “grouped gRNA” is tested for association against the responses to which the element is paired.grna_integration_strategyalso can be set to “singleton,” in which case each gRNA targeting a given element is tested individually against the responses paired to that element. In our analysis we use the default “union” strategy.
Finally, we set the analysis parameters by calling the function
set_analysis_parameters(), passing
sceptre_object, discovery_pairs,
positive_control_pairs, and side as arguments.
Note that sceptre_object is the only required arguments to
this function.
sceptre_object <- set_analysis_parameters(
sceptre_object = sceptre_object,
discovery_pairs = discovery_pairs,
positive_control_pairs = positive_control_pairs,
side = side
)
print(sceptre_object) # output suppressed for brevity3. Assign gRNAs to cells (optional)
The third step is to assign gRNAs to cells. This step can be skipped,
in which case gRNAs are assigned to cells automatically using default
options. The gRNA assignment step involves using the gRNA UMI counts to
determine which cells contain which gRNAs. We begin by plotting the UMI
count distribution of several randomly selected gRNAs via a call to the
function plot_grna_count_distributions().
plot_grna_count_distributions(sceptre_object)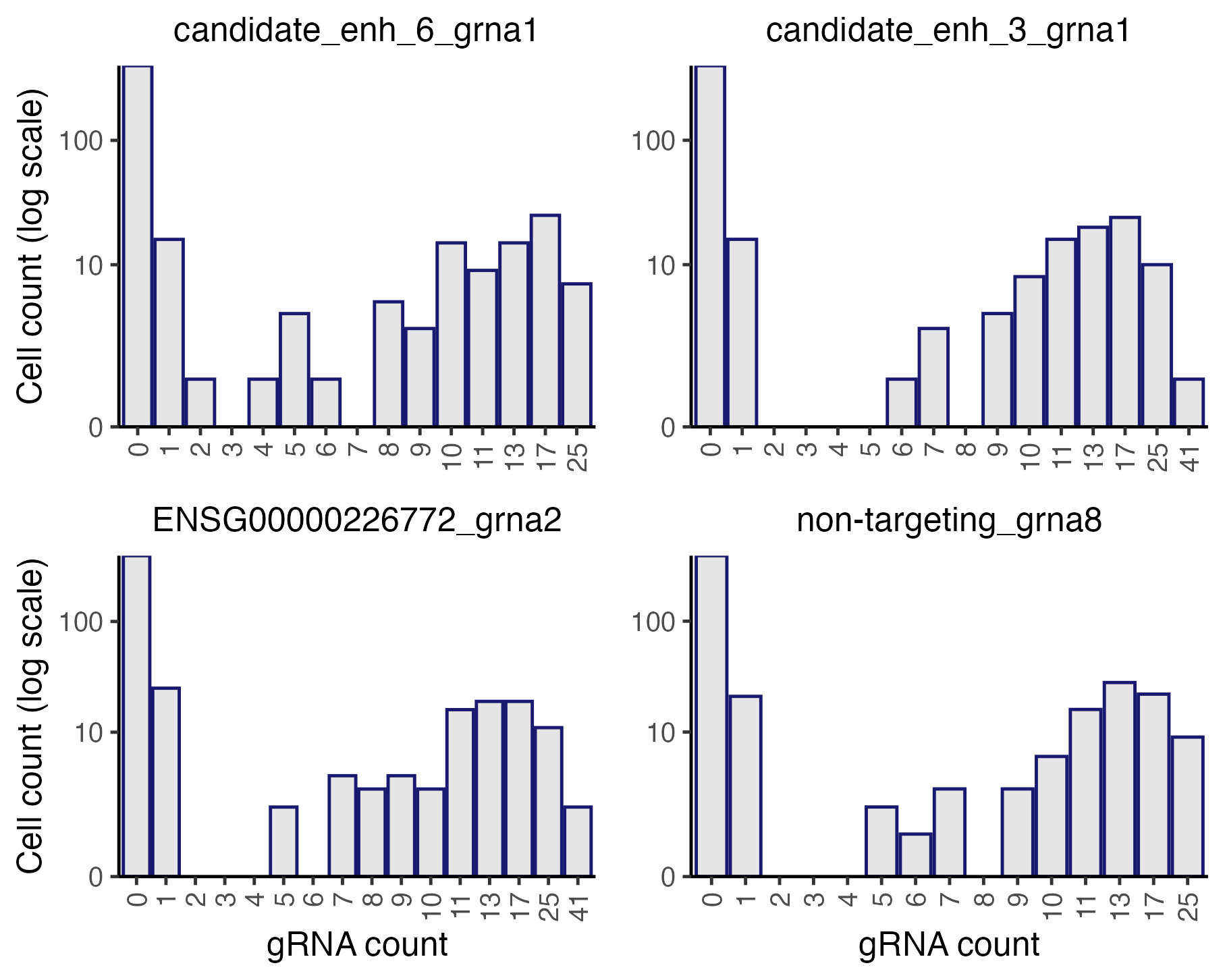
Histograms of the gRNA count distributions
The gRNAs display bimodal count distributions. Consider, for example,
candidate_enh_6_grna1 (top left corner). This gRNA exhibits
a UMI count of
or
in most cells and a UMI count in between in only a handful of cells. The
vast majority of cells with a UMI count of 1 or 2 likely do not actually
contain candidate_enh_6_grna1. This is an example of
“background contamination,” the phenomenon by which gRNA transcripts
sometimes map to cells that do not contain the corresponding gRNA.
sceptre provides three methods for assigning
gRNAs to cells (the “mixture method,” the “maximum method,” and the
“thresholding method”), all of which account for background
contamination. The default method for high-MOI data is the “mixture
method.” The gRNA counts are regressed onto the (unobserved) gRNA
presence/absence indicator and the cell-specific covariates (e.g.,
grna_n_umis, batch) via a latent variable
Poisson GLM. The fitted model yields the probability that each cell
contains the gRNA, and these probabilities are thresholded to assign the
gRNA to cells. The default method in low-MOI is the simpler “maximum”
approach: the gRNA that accounts for the greatest number of UMIs in a
given cell is assigned to that cell. A backup option in both low- and
high-MOI is the “thresholding” approach: a given gRNA is assigned to a
given cell if the UMI count of that gRNA in that cell exceeds some
integer threshold.
We carry out the gRNA assignment step via a call to the function
assign_grnas(). assign_grnas() takes arguments
sceptre_object (required) and method
(optional); the latter argument can be set to "mixture",
"maximum", or "thresholding".
sceptre_object <- assign_grnas(sceptre_object = sceptre_object)
print(sceptre_object) # output suppressed for brevityWe can call plot() on the resulting
sceptre_object to render a plot summarizing the output of
the gRNA-to-cell assignment step.
plot(sceptre_object)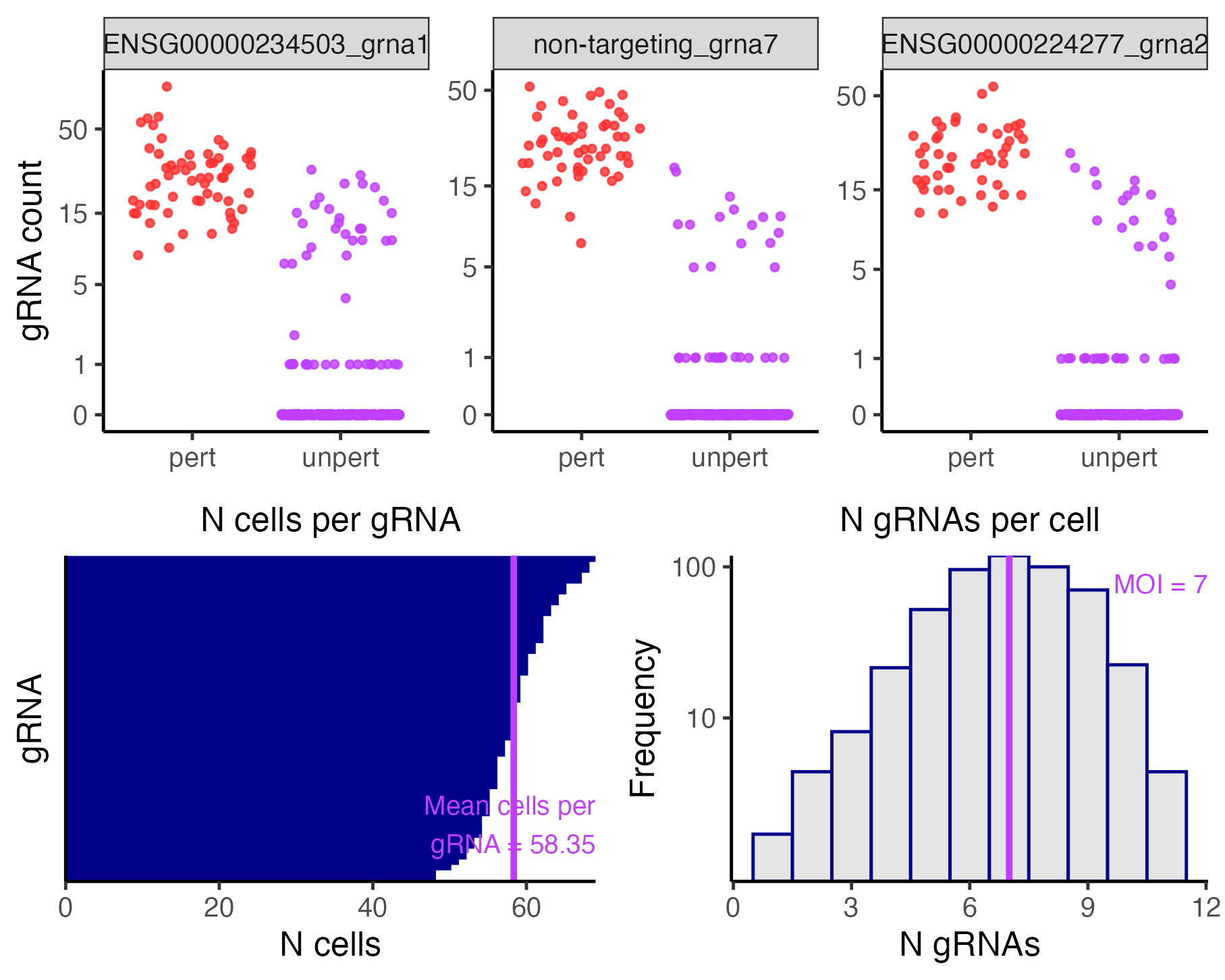
gRNA-to-cell assignments
The top panel plots the gRNA-to-cell assignments of three randomly selected gRNAs. In each plot the points represent cells; the vertical axis indicates the UMI count of the gRNA in a given cell, and the horizontal axis indicates whether the cell has been classified as “perturbed” (i.e., it contains the gRNA) or unperturbed (i.e., it does not contain the gRNA). Perturbed (resp., unperturbed) cells are shown in the left (resp., right) column. The bottom left panel is a barplot of the number of cells to which each gRNA has been mapped. Finally, the bottom right panel is a histogram of the number of gRNAs contained in each cell. The mean number of gRNAs per cell — i.e., the MOI — is displayed in purple text.
4. Run quality control (optional)
The fourth step is to run quality control (QC). This step likewise
can be skipped, in which case QC is applied automatically using default
options. sceptre implements two kinds of QC:
cellwise QC and pairwise QC. The former aims to remove low-quality
cells, while the latter aims to remove low-quality target-response
pairs.
The cellwise QC that sceptre implements is standard in
single-cell analysis. Cells for which response_n_nonzero
(i.e., the number of expressed responses) or
response_n_umis (i.e., the number of response UMIs) are
extremely high or extremely low are removed. Likewise, cells for which
response_p_mito (i.e., the fraction of UMIs mapping to
mitochondrial genes) is excessively high are removed. Additionally, in
low-MOI, cells that contain zero or multiple gRNAs (as determined during
the RNA-to-cell assignment step) are removed. Finally, users optionally
can provide a list of additional cells to remove.
sceptre also implements QC at the level of the
target-response pair. For a given pair we define the “treatment cells”
as those that contain a gRNA targeting the given target. Next, we define
the “control cells” as the cells against which the treatment cells are
compared to carry out the differential expression test. We define the
“number of nonzero treatment cells” (n_nonzero_trt) as the
number of treatment cells with nonzero expression of the
response; similarly, we define the “number of nonzero control cells”
(n_nonzero_cntrl) as the number of control cells
with nonzero expression of the response. sceptre filters
out pairs for which n_nonzero_trt or
n_nonzero_cntrl falls below some threshold (by default
7).
We call the function run_qc() on the
sceptre_object to carry out cellwise and pairwise QC.
run_qc() has several optional arguments that control the
stringency of the various QC thresholds. For example, we set
p_mito_threshold = 0.075, which filters out cells whose
response_p_mito value exceeds 0.075. (The optional
arguments are set to reasonable defaults; the default for
p_mito_threshold is 0.2, for instance).
sceptre_object <- run_qc(sceptre_object, p_mito_threshold = 0.075)
print(sceptre_object) # output suppressed for brevityWe can visualize the output of the QC step by calling
plot() on the updated sceptre_object.
plot(sceptre_object)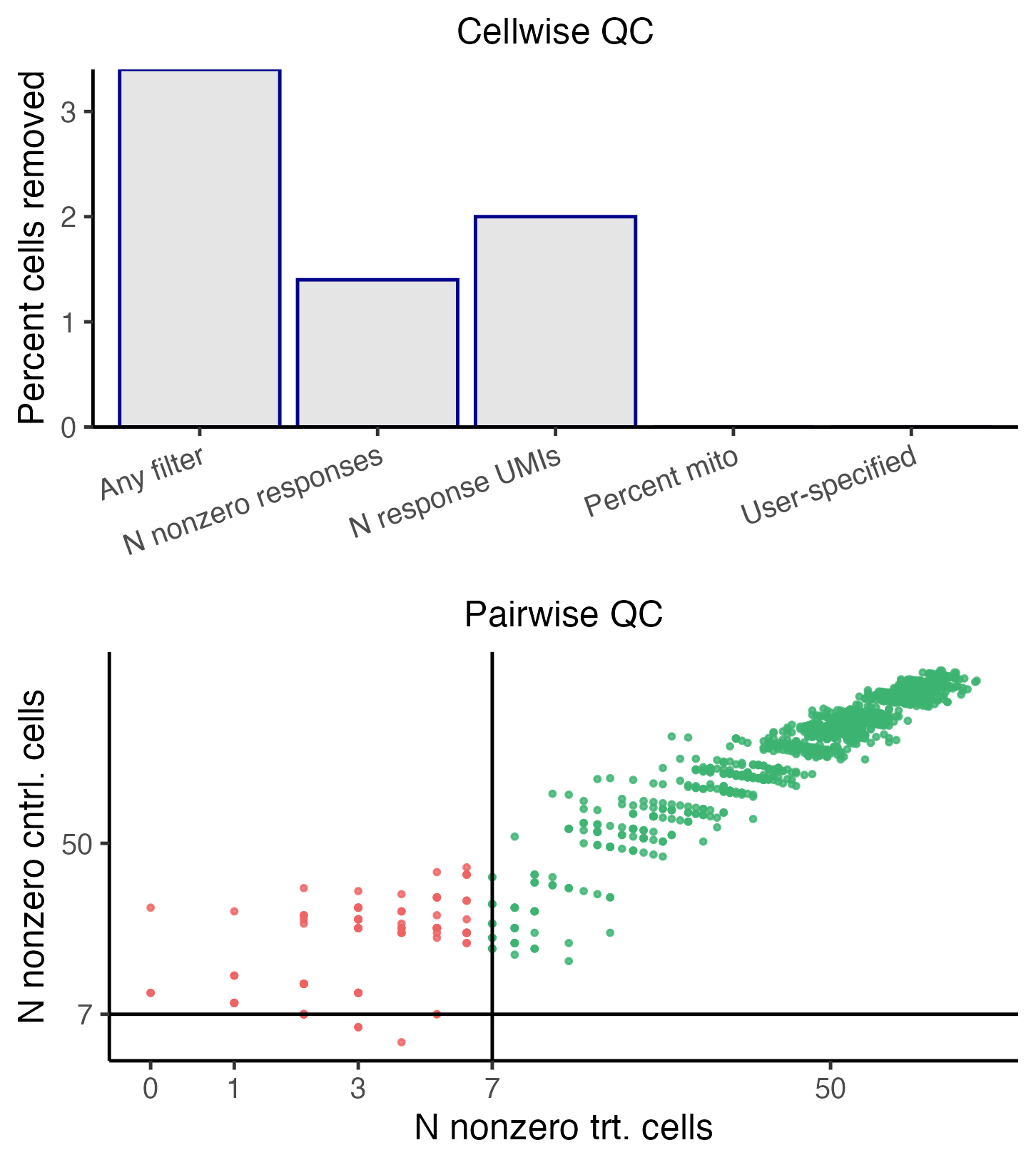
Cellwise and pairwise quality control
The top panel depicts the outcome of the cellwise QC. The various
cellwise QC filters (e.g., “N nonzero responses,” “N response UMIs,”
“Percent mito”, etc.) are shown on the horizontal axis, and the
percentage of cells removed due application of a given QC filter is
shown on the vertical axis. Note that a cell can be flagged by multiple
QC filters; for example, a cell might have an extremely high
response_n_umi value and an extremely high
response_n_nonzero value. Thus, the height of the “any
filter” bar (which indicates the percentage of cells removed due to
application of any filter) need not be equal to the sum of the
heights of the other bars. The bottom panel depicts the outcome of the
pairwise QC. Each point corresponds to a target-response pair; the
vertical axis (resp., horizontal axis) indicates the
n_nonzero_trt (resp., n_nonzero_cntrl) value
of that pair. Pairs for which n_nonzero_trt or
n_nonzero_cntrl fall below the threshold are removed (red),
while the remaining pairs are retained (green).
5. Run calibration check
The fifth step is to run the calibration check. The
calibration check is an analysis that verifies that sceptre
controls the rate of false discoveries on the dataset under
analysis. The calibration check proceeds as follows. First,
negative control target-response pairs are constructed (automatically)
by coupling subsets of NT gRNAs to randomly selected responses.
Importantly, the negative control pairs are constructed in such a way
that they are similar to the discovery pairs, the difference being that
the negative control pairs are devoid of biological signal. Next,
sceptre is applied to analyze the negative control pairs.
Given that the negative control pairs are absent of signal,
sceptre should produce approximately uniformly distributed
p-values on the negative control pairs. Moreover, after an appropriate
multiple testing correction, sceptre should make zero (or
very few) discoveries on the negative control pairs. Verifying
calibration via the calibration check increases our confidence that the
discovery set that sceptre ultimately produces is
uncontaminated by excess false positives.
We run the calibration check by calling the function
run_calibration_check() on the
sceptre_object.
sceptre_object <- run_calibration_check(sceptre_object)
print(sceptre_object) # output suppressed for brevityWe can assess the outcome of the calibration check by calling
plot() on the resulting sceptre_object.
plot(sceptre_object)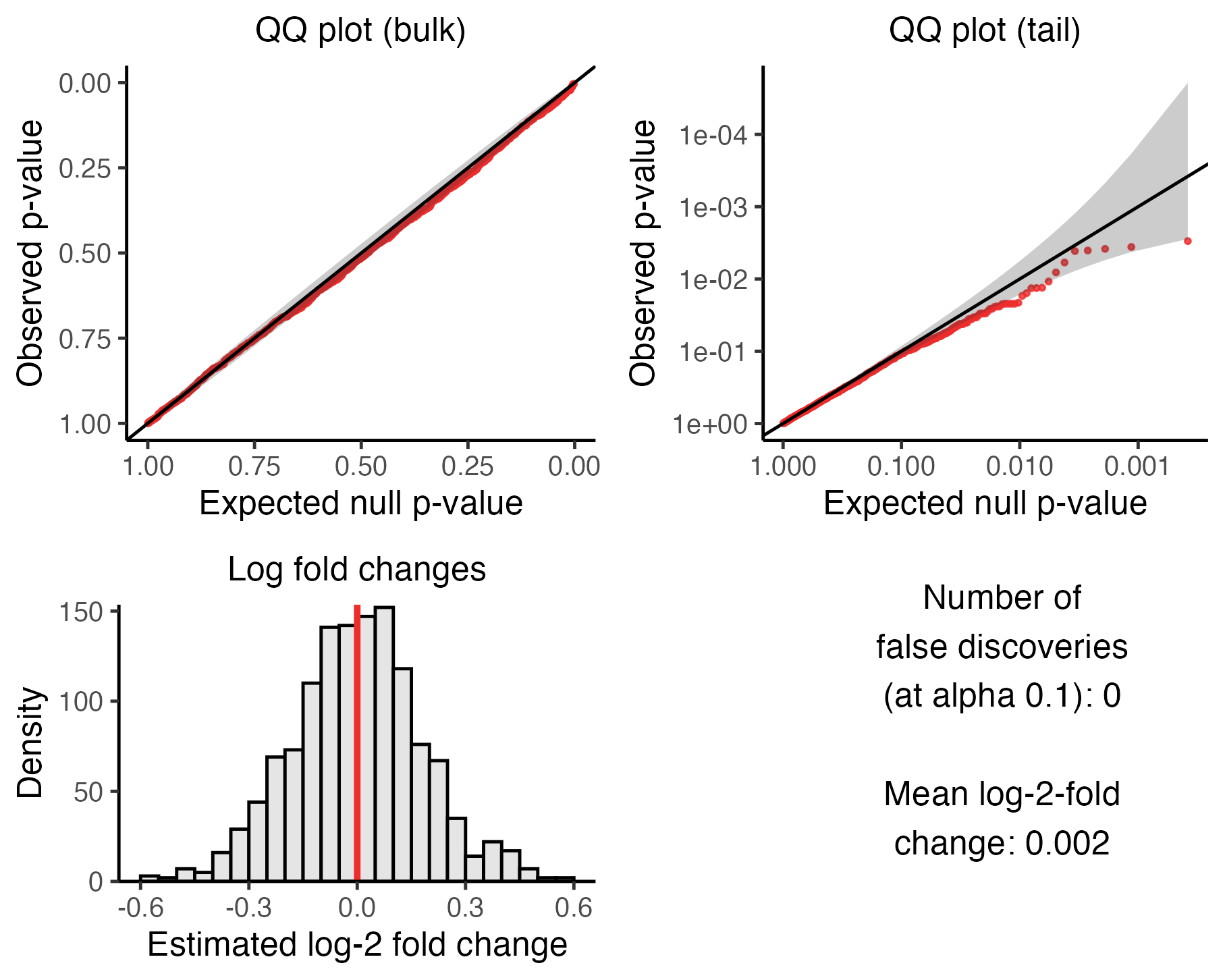
Calibration check results
The visualization consists of four panels, which we describe below.
The upper left panel is a QQ plot of the p-values plotted on an untransformed scale. The p-values should lie along the diagonal line, indicating uniformity of the p-values in the bulk of the distribution.
The upper right panel is a QQ plot of the p-values plotted on a negative log-10 transformed scale. The p-values again should lie along the diagonal line (with the majority of the p-values falling within the gray confidence band), indicating uniformity of the p-values in the tail of the distribution.
The lower left panel is a histogram of the estimated log-2 fold changes. The histogram should be roughly symmetric and centered around zero.
-
Finally, the bottom right panel is a text box displaying (i) the number of false discoveries that
sceptrehas made on the negative control data and- the mean estimated log-fold change. The number of false discoveries should be a small integer like zero, one, two, or three, with zero being ideal. The mean estimated log-fold change, meanwhile, should be a numeric value close to zero; a number in the range [-0.1, 0.1] is adequate.
sceptre may not exhibit good calibration initially,
which is OK. See the book for
strategies for improving calibration.
6. Run power check (optional)
The sixth step — which is optional — is to run the power check. The
power check involves applying sceptre to analyze the
positive control pairs. Given that the positive control pairs are known
to contain signal, sceptre should produce significant
(i.e., small) p-values on the positive control pairs. The power
check enables us to assess sceptre’s power (i.e., its
ability to detect true associations) on the dataset under
analysis. We run the power check by calling the function
run_power_check() on the sceptre_object.
sceptre_object <- run_power_check(sceptre_object)
print(sceptre_object) # output suppressed for brevityWe can visualize the outcome of the power check by calling
plot() on the resulting sceptre_object.
plot(sceptre_object)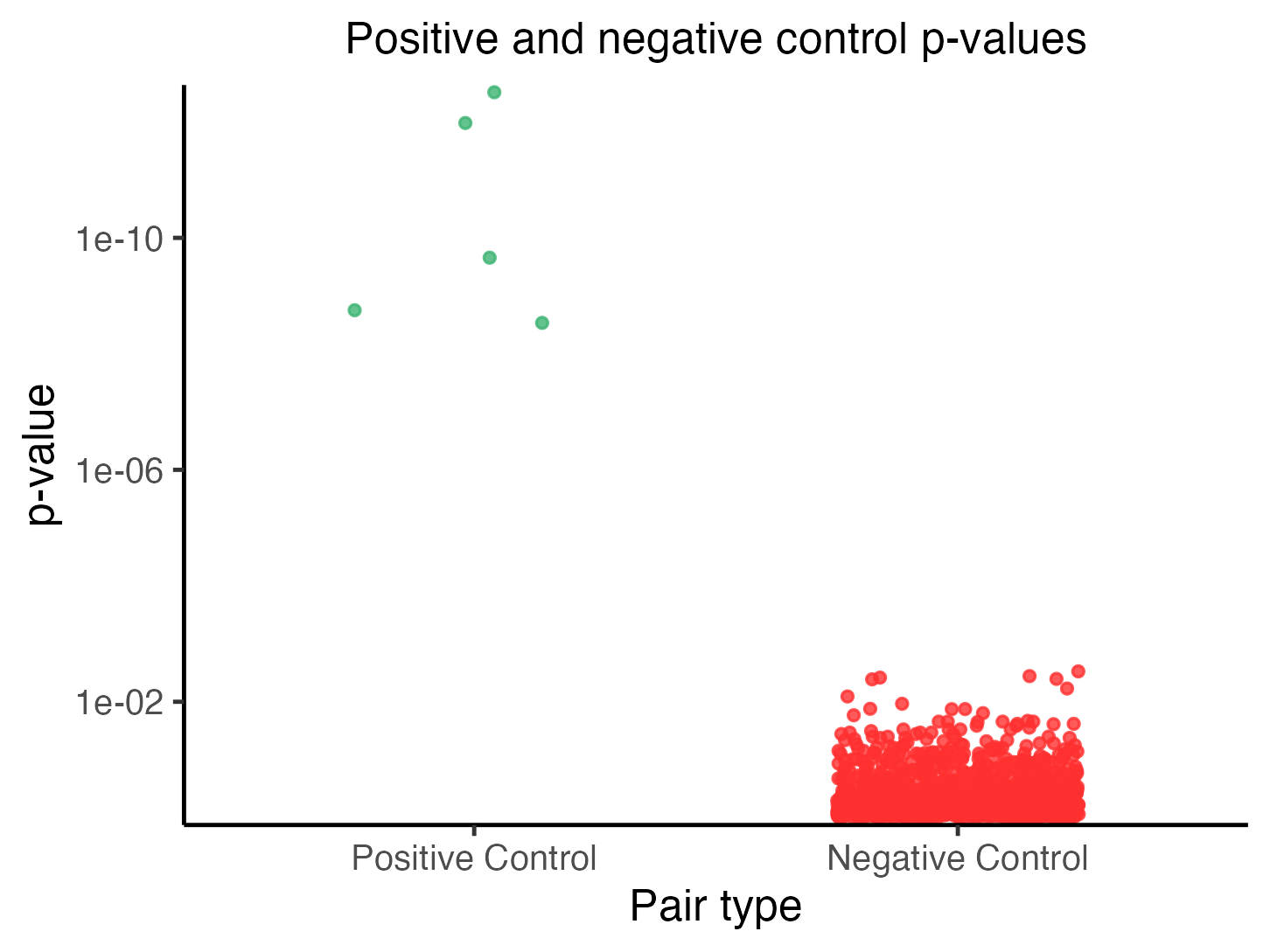
Power check results
Each point in the plot corresponds to a target-response pair, with positive control pairs in the left column and negative control pairs in the right column. The vertical axis indicates the p-value of a given pair; smaller (i.e., more significant) p-values are positioned higher along this axis (p-values truncated at for visualization). The positive control p-values should be small, and in particular, smaller than the negative control p-values.
7. Run discovery analysis
The seventh and penultimate step is to run the discovery analysis.
The discovery analysis entails applying sceptre to analyze
the discovery pairs. We run the discovery analysis by calling the
function run_discovery_analysis().
sceptre_object <- run_discovery_analysis(sceptre_object)
print(sceptre_object) # output suppressed for brevityWe can visualize the outcome of the discovery analysis by calling
plot() on the resulting sceptre_object.
plot(sceptre_object)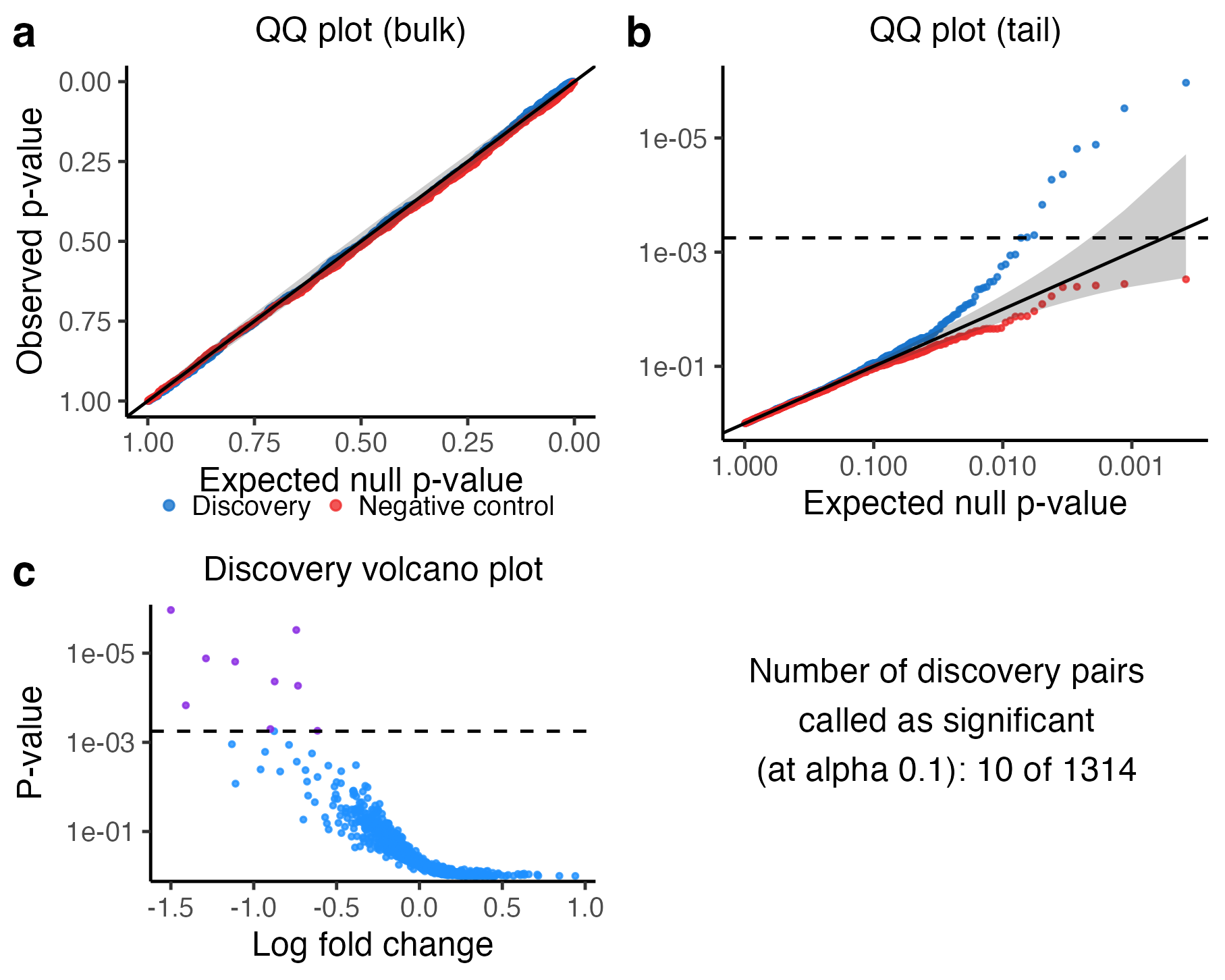
Discovery analysis results
The visualization consists of four panels.
The upper left plot superimposes the discovery p-values (blue) on top of the negative control p-values (red) on an untransformed scale.
The upper right plot is the same as the upper left plot, but the scale is negative log-10 transformed. The discovery p-values should trend above the diagonal line, indicating the presence of signal in the discovery set. The horizontal dashed line indicates the multiple testing threshold; discovery pairs whose p-value falls above this line are called as significant.
The bottom left panel is a volcano plot of the p-values and log fold changes of the discovery pairs. Each point corresponds to a pair; the estimated log-2 fold change of the pair is plotted on the horizontal axis, and the (negative log-10 transformed) p-value is plotted on the vertical axis. The horizontal dashed line again indicates the multiple testing threshold. Points above the dashed line (colored in purple) are called as discoveries, while points below (colored in blue) are called as insignificant.
The bottom right panel is a text box displaying the number of discovery pairs called as significant.
8. Write outputs to directory
The eighth and final step is to write the outputs of the analysis to
a directory on disk. We call the function
write_outputs_to_directory(), which takes as arguments a
sceptre_object and directory;
directory is a string indicating the location of the
directory in which to write the results contained within the
sceptre_object.
write_outputs_to_directory(
sceptre_object = sceptre_object,
directory = output_directory
)write_outputs_to_directory() writes several files to the
specified directory: a text-based summary of the analysis
(analysis_summary.txt), the various plots
(*.png), the calibration check, power check, discovery
analysis results (results_run_calibration_check.rds,
results_run_power_check.rds, and
results_run_discovery_analysis.rds, respectively), and the
binary gRNA-to-cell assignment matrix
(grna_assignment_matrix.rds).
list.files(output_directory)## [1] "analysis_summary.txt" "grna_assignment_matrix.rds"
## [3] "plot_assign_grnas.png" "plot_grna_count_distributions.png"
## [5] "plot_run_calibration_check.png" "plot_run_discovery_analysis.png"
## [7] "plot_run_power_check.png" "plot_run_qc.png"
## [9] "results_run_calibration_check.rds" "results_run_discovery_analysis.rds"
## [11] "results_run_power_check.rds"We also can obtain the calibration check, power check, and discovery
analysis results in R via a call to the function
get_result(), passing as arguments
sceptre_object and analysis, where the latter
is a string indicating the function whose results we are querying.
result <- get_result(
sceptre_object = sceptre_object,
analysis = "run_discovery_analysis"
)The variable result is a data frame, the rows of which
correspond to target-response pairs, and the columns of which are as
follows: response_id, grna_target,
n_nonzero_trt, n_nonzero_cntrl,
pass_qc (a TRUE/FALSE value
indicating whether the pair passes pairwise QC), p_value,
fold_change, se_fold_change (standard error
for fold change estimate), log_2_fold_change, and
significant (a TRUE/FALSE value
indicating whether the pair is called as significant). The p-value
contained within the p_value column is a raw (i.e.,
non-multiplicity-adjusted) p-value.
head(result)## response_id grna_target n_nonzero_trt n_nonzero_cntrl pass_qc
## <char> <char> <int> <int> <lgcl>
## 1: ENSG00000220891 candidate_enh_17 20 169 TRUE
## 2: ENSG00000211641 candidate_enh_15 77 301 TRUE
## 3: ENSG00000220891 candidate_enh_18 22 167 TRUE
## 4: ENSG00000211655 candidate_enh_5 29 165 TRUE
## 5: ENSG00000253451 candidate_enh_19 45 227 TRUE
## 6: ENSG00000211641 candidate_enh_16 75 303 TRUE
## p_value fold_change se_fold_change log_2_fold_change significant
## <num> <num> <num> <num> <lgcl>
## 1: 1.076683e-06 0.3524393 0.07719320 -1.5045534 TRUE
## 2: 3.021114e-06 0.5974147 0.06777816 -0.7431952 TRUE
## 3: 1.311570e-05 0.4094443 0.08408239 -1.2882609 TRUE
## 4: 1.550816e-05 0.4626207 0.08963478 -1.1120984 TRUE
## 5: 4.323042e-05 0.5457610 0.07300526 -0.8736588 TRUE
## 6: 5.376764e-05 0.6016546 0.06982219 -0.7329927 TRUEFurther reading
We encourage readers interested in learning more to consult the sceptre manual.
## ─ Session info ───────────────────────────────────────────────────────────────
## setting value
## version R version 4.6.0 (2026-04-24)
## os macOS Tahoe 26.4.1
## system aarch64, darwin23
## ui X11
## language en
## collate en_US
## ctype en_US
## tz America/New_York
## date 2026-06-24
## pandoc 3.9.0.2 @ /opt/homebrew/bin/ (via rmarkdown)
## quarto 1.9.38 @ /usr/local/bin/quarto
##
## ─ Packages ───────────────────────────────────────────────────────────────────
## package * version date (UTC) lib source
## bslib 0.10.0 2026-01-26 [3] CRAN (R 4.6.0)
## cachem 1.1.0 2024-05-16 [3] CRAN (R 4.6.0)
## cli 3.6.6 2026-04-09 [3] CRAN (R 4.6.0)
## cowplot 1.2.0 2025-07-07 [3] CRAN (R 4.6.0)
## crayon 1.5.3 2024-06-20 [3] CRAN (R 4.6.0)
## data.table 1.18.4 2026-05-06 [3] CRAN (R 4.6.0)
## desc 1.4.3 2023-12-10 [3] CRAN (R 4.6.0)
## digest 0.6.39 2025-11-19 [3] CRAN (R 4.6.0)
## dplyr 1.2.1 2026-04-03 [3] CRAN (R 4.6.0)
## evaluate 1.0.5 2025-08-27 [3] CRAN (R 4.6.0)
## farver 2.1.2 2024-05-13 [3] CRAN (R 4.6.0)
## fastmap 1.2.0 2024-05-15 [3] CRAN (R 4.6.0)
## fs 2.1.0 2026-04-18 [3] CRAN (R 4.6.0)
## generics 0.1.4 2025-05-09 [3] CRAN (R 4.6.0)
## ggplot2 4.0.3 2026-04-22 [3] CRAN (R 4.6.0)
## glue 1.8.1 2026-04-17 [3] CRAN (R 4.6.0)
## gtable 0.3.6 2024-10-25 [3] CRAN (R 4.6.0)
## htmltools 0.5.9 2025-12-04 [3] CRAN (R 4.6.0)
## htmlwidgets 1.6.4 2023-12-06 [3] CRAN (R 4.6.0)
## jquerylib 0.1.4 2021-04-26 [3] CRAN (R 4.6.0)
## jsonlite 2.0.0 2025-03-27 [3] CRAN (R 4.6.0)
## knitr 1.51 2025-12-20 [3] CRAN (R 4.6.0)
## labeling 0.4.3 2023-08-29 [3] CRAN (R 4.6.0)
## lattice 0.22-9 2026-02-09 [3] CRAN (R 4.6.0)
## lifecycle 1.0.5 2026-01-08 [3] CRAN (R 4.6.0)
## magrittr 2.0.5 2026-04-04 [3] CRAN (R 4.6.0)
## Matrix * 1.7-5 2026-03-21 [3] CRAN (R 4.6.0)
## otel 0.2.0 2025-08-29 [3] CRAN (R 4.6.0)
## pillar 1.11.1 2025-09-17 [3] CRAN (R 4.6.0)
## pkgconfig 2.0.3 2019-09-22 [3] CRAN (R 4.6.0)
## pkgdown 2.2.0 2025-11-06 [3] CRAN (R 4.6.0)
## purrr 1.2.2 2026-04-10 [3] CRAN (R 4.6.0)
## R.methodsS3 1.8.2 2022-06-13 [3] CRAN (R 4.6.0)
## R.oo 1.27.1 2025-05-02 [3] CRAN (R 4.6.0)
## R.utils 2.13.0 2025-02-24 [3] CRAN (R 4.6.0)
## R6 2.6.1 2025-02-15 [3] CRAN (R 4.6.0)
## ragg 1.5.2 2026-03-23 [3] CRAN (R 4.6.0)
## RColorBrewer 1.1-3 2022-04-03 [3] CRAN (R 4.6.0)
## Rcpp 1.1.1-1.1 2026-04-24 [3] CRAN (R 4.6.0)
## rlang 1.2.0 2026-04-06 [3] CRAN (R 4.6.0)
## rmarkdown 2.31 2026-03-26 [3] CRAN (R 4.6.0)
## S7 0.2.2 2026-04-22 [3] CRAN (R 4.6.0)
## sass 0.4.10 2025-04-11 [3] CRAN (R 4.6.0)
## scales 1.4.0 2025-04-24 [3] CRAN (R 4.6.0)
## sceptre * 0.99.0 2026-06-24 [1] Bioconductor
## sessioninfo * 1.2.3 2025-02-05 [3] CRAN (R 4.6.0)
## systemfonts 1.3.2 2026-03-05 [3] CRAN (R 4.6.0)
## textshaping 1.0.5 2026-03-06 [3] CRAN (R 4.6.0)
## tibble 3.3.1 2026-01-11 [3] CRAN (R 4.6.0)
## tidyselect 1.2.1 2024-03-11 [3] CRAN (R 4.6.0)
## utf8 1.2.6 2025-06-08 [3] CRAN (R 4.6.0)
## vctrs 0.7.3 2026-04-11 [3] CRAN (R 4.6.0)
## withr 3.0.2 2024-10-28 [3] CRAN (R 4.6.0)
## xfun 0.57 2026-03-20 [3] CRAN (R 4.6.0)
## yaml 2.3.12 2025-12-10 [3] CRAN (R 4.6.0)
##
## [1] /private/var/folders/1w/h831hyps5qs5lzkh5xjj0_wh0000gq/T/RtmpgcetiN/temp_libpath124931c84cecb
## [2] /Users/ekatsevi/Library/R/arm64/4.6/library
## [3] /Library/Frameworks/R.framework/Versions/4.6/Resources/library
## * ── Packages attached to the search path.
##
## ──────────────────────────────────────────────────────────────────────────────